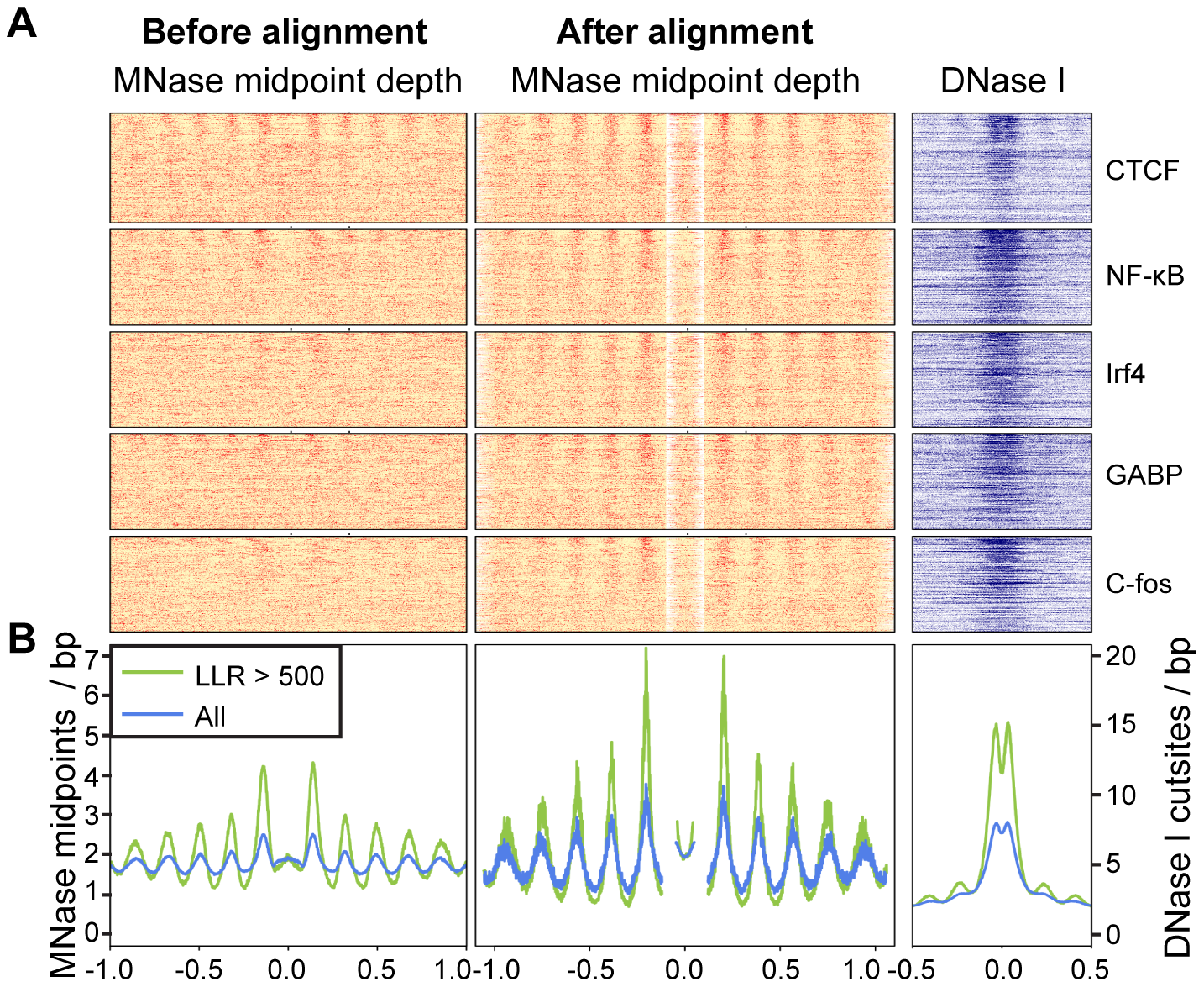
# A function to reorder the rows of a matrix according to the overall
# similarity (correlation) of each row to the column sums of the matrix.
# The top rows will have the highest similarity with the column sums and
# the bottom rows the lowest.
# MATRIX a matrix of numerics of interest
# Returns the same matrix with the rows reordered
order.rows = function(MATRIX)
{ REF = colSums(MATRIX)
SCORES = apply(MATRIX, 1, cor, REF)
ord = order(SCORES, decreasing=F)
return(MATRIX[ord,])
}
# plot
# install package 'zoo' using if needed
# install.packages("zoo")
library(zoo)
color = colorRampPalette(c("white", "red"), space = "rgb")(100)
X = rollapply(order.rows(DATA), width=20, mean, by=20, by.row=TRUE)
Y = rollapply(order.rows(DATA.aligned), width=20, mean, by=20, by.row=TRUE)
layout(matrix(c(1,2,3,4), nrow=2, ncol=2, byrow=T), heights=c(1.5,1.5))
image(t(X), col=color, xaxt="n", yaxt="n", bty="n")
title("Before alignment")
image(t(Y), col=color, xaxt="n", yaxt="n", bty="n")
title("After alignment")
plot(seq(-990, 990, 10), colMeans(DATA), type="l", lwd=2, ylab="", xlab="", bty="n", ylim=c(0,12))
plot(seq(-990, 990, 10), colMeans(DATA.aligned), type="l", lwd=2, ylab="", xlab="", bty="n", ylim=c(0,12))
plot(seq(-990, 990, 10), colSums(DATA.aligned), type="l", lwd=2, ylab="", xlab="", bty="n",col="green", ylim=c(0,1.7))