Chip-seq data analysis: from quality check to motif discovery and more
Lausanne, 27 March - 31 March 2017
Data reproduction exercise: Variation in Drosophila transcription factor binding sites.
Author: Philipp Bucher
Introduction
This exercise is based on the following paper:
Spivakov et al. 2012.
Analysis of variation at transcription factor binding sites in Drosophila and humans.
Genome Biol. 13:R49.
PMID: 22950968
The authors analyze cross-species and intra-species variation of transcription factor
binding sites identified in previous studies. We will try to reproduce some of the results
concerning bindings sites for the Drosophila transcription factors Twist, Binou and Tinman reported
in:
Zinzen et al. 2009.
Combinatorial binding predicts spatio-temporal cis-regulatory activity.
Nature 462, 65-70.
PMID: 19890324
These binding sites were identified by scanning ChIP-on-chip peak regions with newly derived PWMs for the
transcription factor under investigation.
Exercise
We wil try to reprocude results shown in Figure 1a,b in
(Spivakov et al. 2012).
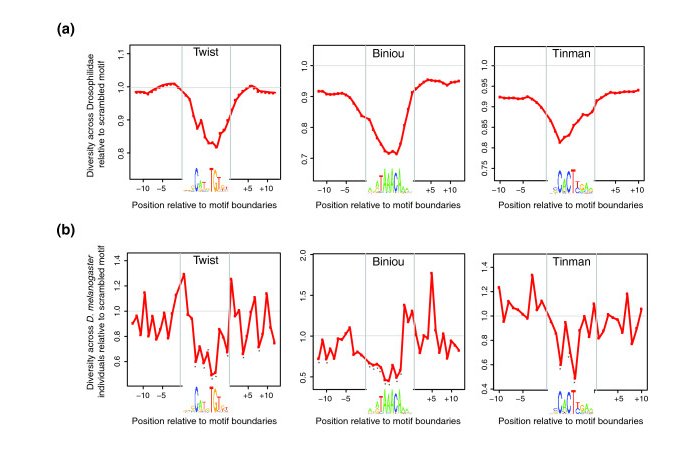
Have a look at the Figure legend and the Methods section of the corresponding paper.
However, you are not asked to precisely follow the data analysis protocol of the others. Just use
the relevant sequence conservation and SNP tracks from the ChIP-Seq server to produce
figures of the same kind. Once you have the results, you may ask yourself whether you agree
with the interpretation by the authors:
"As expected, TF-bound motifs both are conserved across evolutionary distance and
show depressed levels of variation across individuals compared to either their
respective flanking regions ..."
The genomic coordinates upon which this analysis is based are given in supplementary
Table S3. Note the Figure 1a is based in PhastCons scores. Higher resolution pictures
could potentially be obtainied using the PhyloP track, which was probably not yet
available when the paper was published.
Hints and recipes
The supplementary Table S3 is a tab-delimited text file which
provides binding sites coordinates for several transcription
factors in the following format.
CHR START END STRAND SEQ TF PWM_SCORE ...
2L 18564 18574 1 TAGTGTGCCCG trx_disc1 6.06 ...
2L 18917 18927 1 CAGTGTGACCA trx_disc1 8.2 ...
2L 21369 21379 1 TAGTGTGCCCG trx_disc1 6.06 ...
2L 21742 21752 1 CAGTGTGACCA trx_disc1 8.2 ...
2L 22068 22078 1 CAGTGTGGACG trx_disc1 6.15 ...
2L 28228 28237 -1 AGCTACCTGT hb_disc1 6.16 ...
The genomic coordinates refer to the D. melanogaster genome assembly dm3.
The file contains binding sites for 10 different transcription factors identified
by the codes:
bin_ef, cnc_disc1, h_known1, hb_disc1, hkb_known2, mod_disc3, tin_ef, trx_disc1
and twi_ef- We are only interested in
twi_ef (Twist), bin_ef (Binou) and tin_ef (Tinman).
Several editing operations need be applied to transform the lines of this
table into a valid BED file that can be uploaded to the ChIP-seq server:
- The chromosome identifier should be made USCS-style, e.g. 2L -> chr2L.
- The start coordinate is 1-based in Table S3. It should be 0-based for BED.
- The strand field should be either "+" or "-" and it should appear in the 6th column.
The following R code could be used to extract appropriate binding site lists.
bed = read.table("spivakov12_S3.txt", as.is=1, header=T)
bed[,1]=paste("chr", as.character(bed[,1]), sep="")
bed[,2]=bed[,2]-1
bed[which(bed[,4] == 1),4] = "+"
bed[which(bed[,4] == -1),4] = "-"
write.table(bed[which(bed[,6] == "twi_ef"),c(1,2,3,5,6,4)],
"twi_ef.bed", quote=F, sep="\t", row.names=F, col.names=F)
write.table(bed[which(bed[,6] == "tin_ef"),c(1,2,3,5,6,4)],
"tin_ef.bed", quote=F, sep="\t", row.names=F, col.names=F)
write.table(bed[which(bed[,6] == "bin_ef"),c(1,2,3,5,6,4)],
"bin_ef.bed", quote=F, sep="\t", row.names=F, col.names=F)
The bed files produced in this way can be uploaded to
ChIP-Convert,
conversion options:
BED-to-SGA, compress output=off, Genome=D. melanogaster (Apr 2006 R5/dm3)
All other switches should be left blank.
From the results page, you can directly navigate to the ChIP-Cor server.
Since the binding motifs are asymmetric and the binding sites may
occur in either orientation, you should specify "strand oriented" for
the reference feature. The count cut-off may invariantly be set to 10
since as all relevant tracks have count values of at most 10.
Relevant target feature tracks for this exercise are:
- Sequence-derived -> SNP collections from dbSNP -> Genomic SNPs
- Sequence-derived -> PhastCons 15-way ... -> PHASTCONS 15-way
- Sequence-derived -> Phylop basewise conservation -> PhyloP (27 species) score >= 2