Lausanne, 27 March - 31 March 2017
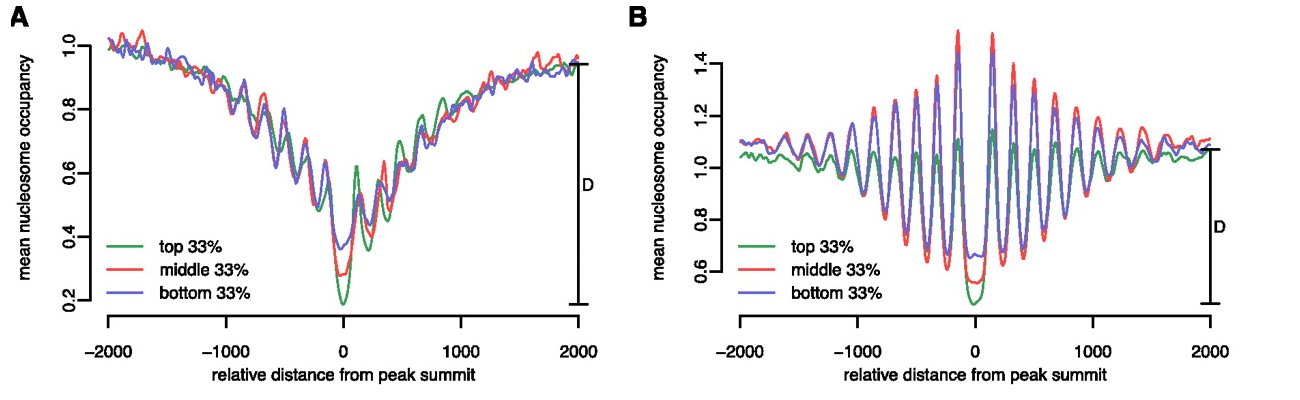
The Figure shows average nucleosome occupancy profiles around YY1 binding sites in the lymphoblastoid cell line GM12878. The peak lists were first divided into promoter-proximal and promoter-distal binding sites and further stratified into three groups based on ChIP-seq signal strength.
The authors of the paper describe and interpret the results as follows:
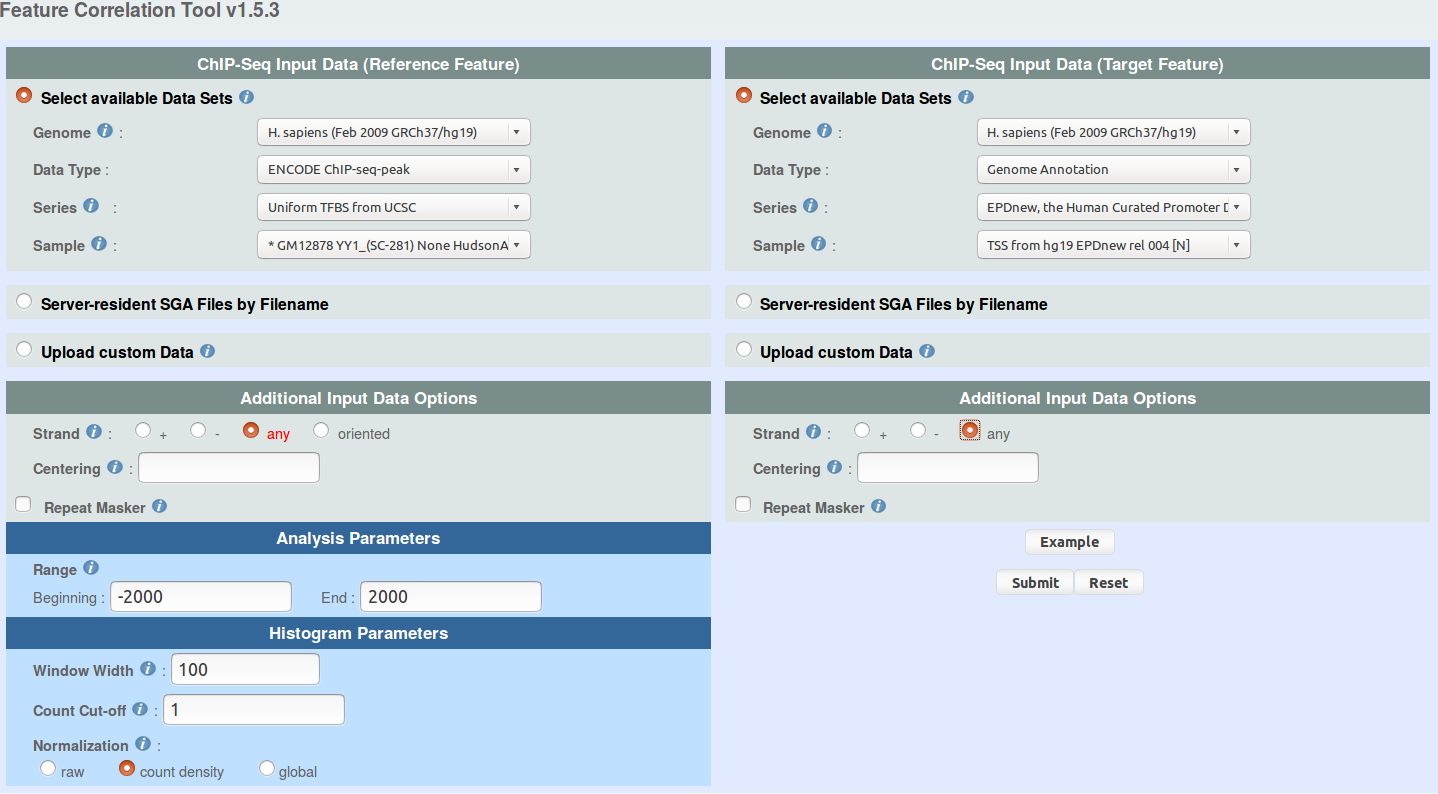
ENCODE ChIP-seq-peak -> Uniform TFBS from UCSC
sample: GM12878 YY1_(SC-281) None HudsonAlpha peaks
ENCODE DNase FAIRE etc. -> GSE35586, Nucleosome Position by MNase-seq ...
sample: Nucleosome Gm12878 rep1
Genome Annotation -> EPD, the Human Curated Promoter Database
sample: TSS from hg19 EPDnew rel 003
Note: The MNase data files are very voluminous and thus take time to process
(minutes rather than secons). We therefore recommend to generate the figures
with one replicate only.
Making a nucleosome occupancy profile for distal YY1 sites.
In the paper, the authors write:
Stratifying the YY1 peaks into three subsets by signal strength
One way of doing this is by downloading the peak SGA file from the MGA repository via the MGA overview page at:
sga=read.table("wgEncodeAwgTfbsHaibGm12878Yy1sc281Pcr1xUniPk.sga.gz", header=F)
dim(sga)
N=dim(sga)[1]
r1 = 1:floor(N/3)
r2 = (floor(N/3)+1):floor(2*N/3)
r3 = (floor(2*N/3)+1):N
bottom = sort(order(sga[,6])[r1])
middle = sort(order(sga[,6])[r2])
top = sort(order(sga[,6])[r3])
write.table(sga[bottom,], "GM12878_YY1_bottom.sga",
quote=F, sep="\t", row.names=F, col.names=F)
write.table(sga[middle,], "GM12878_YY1_middle_sga",
quote=F, sep="\t", row.names=F, col.names=F)
write.table(sga[top,], "GM12878_YY1_top.sga",
quote=F, sep="\t", row.names=F, col.names=F)
You can now individually upload these SGA files to the
ChIP-cor server
and repeat the previously outlined procedure to generate
nucleosome occupancy plots.
A more efficient prodedures to reproduc Figures 5a and 5b at once
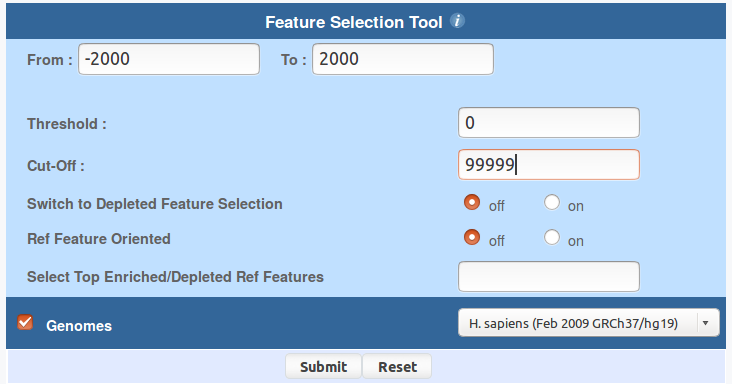
You can use ChIP-Cor to generate an extended YY1 peak file with promoter status annotation. Select the above indicated YY1 peak as reference feature and EPD promoters as target feature. Have a look at the following pictures to set the parameters. In essence, the use of the "Enriched Feature Extraction Option" with Threshold=0 will result in selecting all the promoters. However, you will know the number of TSS beside every peak. Save the resulting SGA file under the name GM12878_YY1_anno.sga (or get it here).
NC_000001.10 YY1_P 714001 0 1 133.535942201664 0 NC_000001.10 YY1_P 805331 0 1 46.6896250548417 0 NC_000001.10 YY1_P 840144 0 1 70.0879017887289 0 NC_000001.10 YY1_P 894612 0 1 244.724808404057 2 NC_000001.10 YY1_P 895930 0 1 40.8279820108751 2You can now upload this file to the ChIP-Extract input form at:
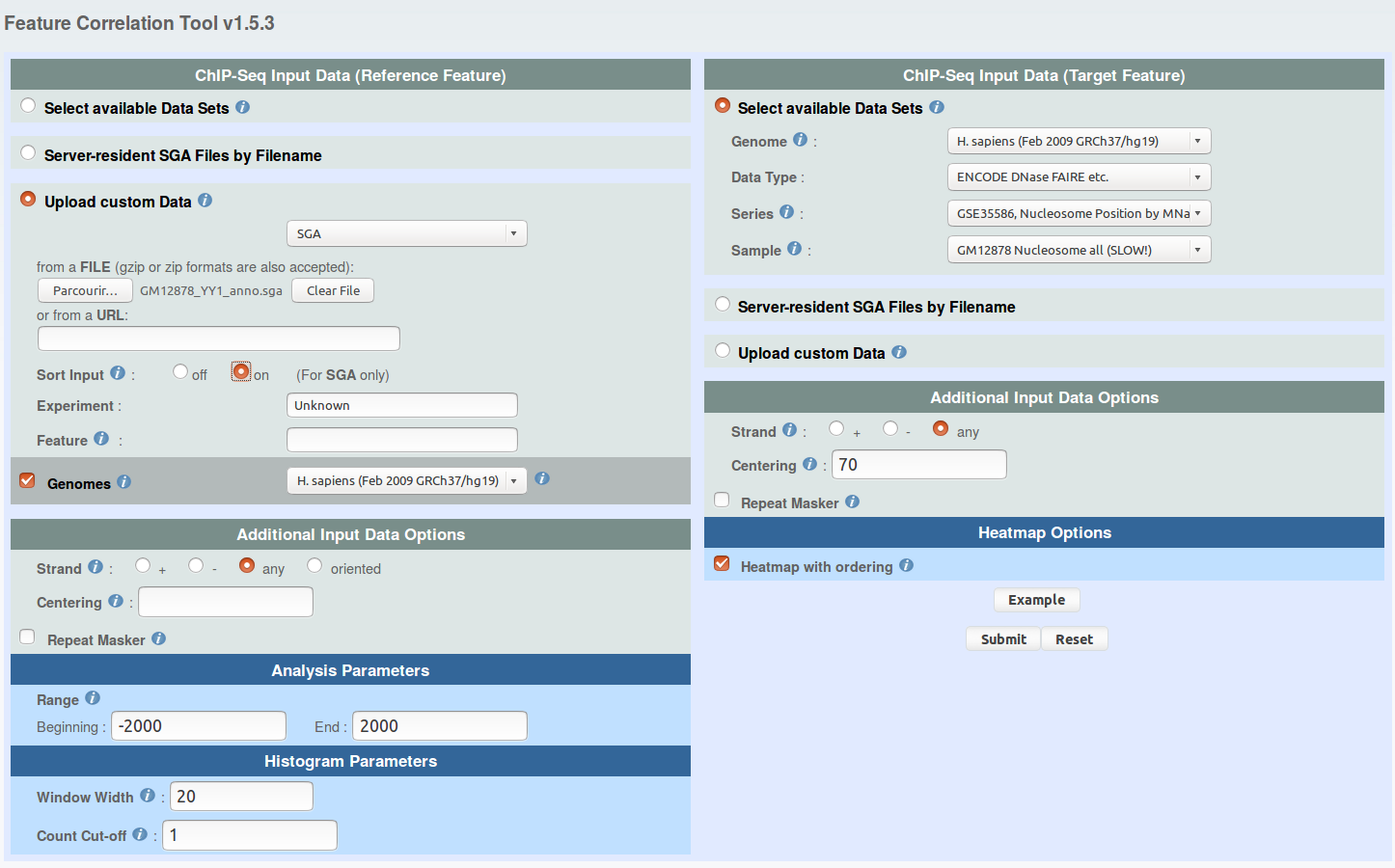
From the ChIP-Extract results page, download the "Ref SGA File" and the "Table (TEXT)" under the names:
Below is the R recipe to generate the plots equivalent to Figure 5a and 5b in the paper.
# data input, defining the proximal and distal peak subsets
anno=read.table("GM12878_YY1_anno.sga")
data=as.matrix(read.table("GM12878_YY1_nucl.txt"))
prox=which(anno[,7] != 0)
dist=which(anno[,7] == 0)
# top, middle, bottom subsets for proximal and distal peaks
N = length(prox)
r1 = 1:floor(N/3)
r2 = (floor(N/3)+1):floor(2*N/3)
r3 = (floor(2*N/3)+1):N
prox_bottom = prox[sort(order(anno[prox,6])[r1])]
prox_middle = prox[sort(order(anno[prox,6])[r2])]
prox_top = prox[sort(order(anno[prox,6])[r3])]
N = length(dist)
r1 = 1:floor(N/3)
r2 = (floor(N/3)+1):floor(2*N/3)
r3 = (floor(2*N/3)+1):N
dist_bottom = dist[sort(order(anno[dist,6])[r1])]
dist_middle = dist[sort(order(anno[dist,6])[r2])]
dist_top = dist[sort(order(anno[dist,6])[r3])]
layout(matrix(1:2, nrow=1, ncol=2))
# plotting Figure 5a
x = 20*(-99:99); par(cex=1.2)
plot(x,colMeans(data[prox_top,]),t="l", col="green", ylim=c(1.5,6),
xlab="relative distance to peak summit",
ylab="mean nucleosome occupancy")
par(new=T)
plot(x,colMeans(data[prox_middle,]),t="l", col="red", ylim=c(1.5,6),
xaxt="n", yaxt="n", xlab="", ylab="")
par(new=T)
plot(x,colMeans(data[prox_bottom,]),t="l", col="blue", ylim=c(1.5,6),
xaxt="n", yaxt="n", xlab="", ylab="")
legend("bottomleft", legend=c("top 33%","middle 33%", "bottom 33%"),
lty=1, col=c("green","red", "blue"), bty="n")
# plotting Figure 5b
x = 20*(-99:99); par(cex=1.2)
plot(x,colMeans(data[dist_top,]),t="l", col="green", ylim=c(4,7.5),
xlab="relative distance to peak summit",
ylab="mean nucleosome occupancy")
par(new=T)
plot(x,colMeans(data[dist_middle,]),t="l", col="red", ylim=c(4,7.5),
xaxt="n", yaxt="n", xlab="", ylab="")
par(new=T)
plot(x,colMeans(data[dist_bottom,]),t="l", col="blue", ylim=c(4,7.5),
xaxt="n", yaxt="n", xlab="", ylab="")
legend("bottomleft", legend=c("top 33%","middle 33%", "bottom 33%"),
lty=1, col=c("green","red", "blue"), bty="n")