Lausanne, 27 March - 31 March 2017
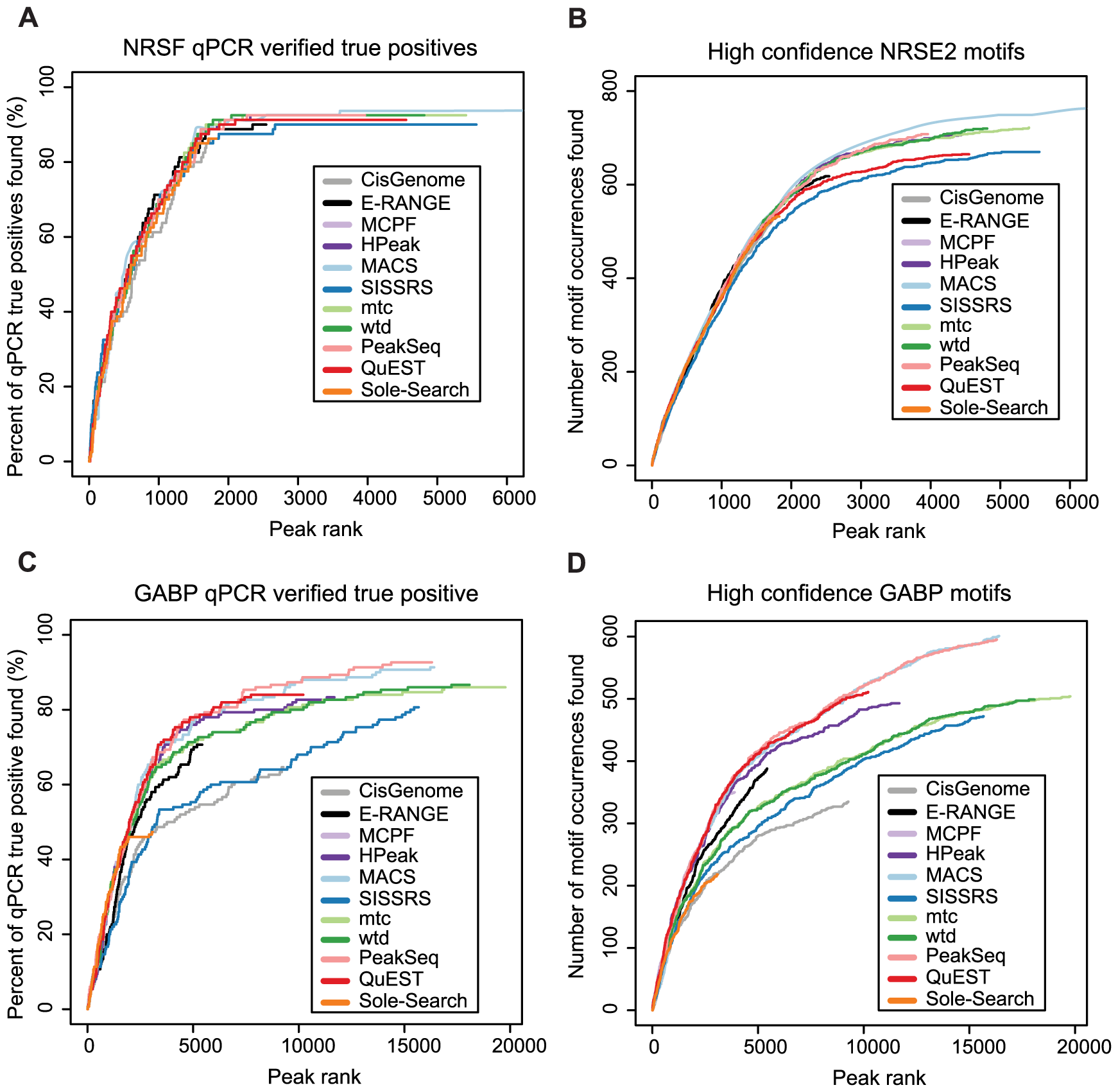
Figure 5 compares peak lists obtained with different peak finders with so-called sensitivity plots.
These plots show the total number of true positive among the N highest ranking peaks as a function of N. True positives are either defined by a given set of experimentally validated binding sites or by the presence of a binding motif of the corresponding transcription factor.
Figure 6 shows the ranking accuracy obtained with different peak finders for NRSF.
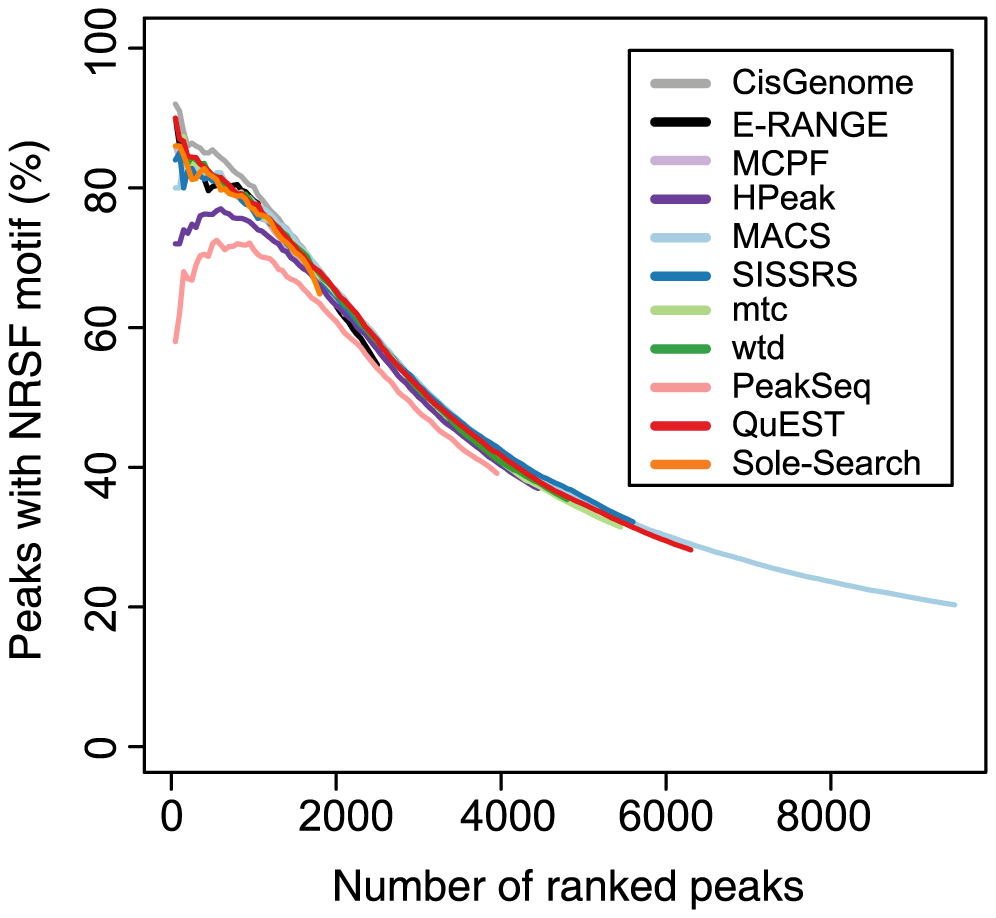
In principle, one would expect the top-ranked peaks to be the most highly enriched in the NRSF binding sites. As you can see, this is not the case for all Peak finders.
ChIP-seq and ChIP-seq-peak data from hg18:
Valouev 2008, Jurkat cells, GABP SRF NRSF
Sample: Jurkat GABP
Sample: Jurkat NRSF(polyclonal)
Sample: Jurkat NRSF(monoclonal)
Additional peak lists for GABP and NRSF from hg19 can be found in:
ENCODE ChIP-seq-peak -> Uniform TFBS from UCSCqPCR-validated binding site lists:
These lists are avaible in a Excel file named "Dataset S1" posted on the publisher's website under "Supporting Information". The Excel file contains two sheets, one for GABP and one for NSRF, which you have to export seperately in Text CSV format (field delimiter tab, text delimiter none). The exported files are posted below:
GABP = read.table("wilbanks_DS1_GABP.txt")
GABP = GABP[-1,]
write.table(GABP[,1:3],"GABP_qPCR.bed",
row.names=F, col.names=F, sep="\t", quote=F)
The binding site table for NRSF has an additional column
filled with the enrichment factor. According to
Mortazavi et al.
(PMID: 16963704)
only sites with enrichment values higher than 2.44
should be considered as positives.
This is taken into account with the following conversion recipe.
NRSF = read.table("wilbanks_DS1_NRSF.txt", as.is=4)
NRSF = NRSF[-1,]
pos=which(as.numeric(NRSF[,4]) > 2.44)
write.table(NRSF[pos,1:3],"NRSF_qPCR.bed",
row.names=F, col.names=F, sep="\t", quote=F)
The BED files can be uploaded to ChIP-Convert
for convervsion into SGA format (Compress output off, centered
SGA on, Unoriented SGA on). We recommend to liftOver the resulting files
to hg19 and to save SGA files for both assemblies.
All files resulting from the conversion procedure are posted below for your convenience. We recommend to use the same file names if you do the conversion yourself.
| GABP_qPCR.bed | GABP_qPCR_hg18.sga | GABP_qPCR_hg19.sga |
| NRSF_qPCR.bed | NRSF_qPCR_hg18.sga | NRSF_qPCR_hg19.sga |
Use the standard procedure proposed in the tutorial of day one. The peak lists below
strand: any Window Width : 200 Count Cut-off: 1 centering: 50 Vicinity Range: 200 Refine Peak Positions: on Repeat Masker: off Peak Threshold: 10Making sensitivity plots with qPCR-validated binding sites
Go to ChIP-Cor. Upload a peak list as reference feature and the corresponding qPCR-validated binding site list as target feature and set the parameters as follows:
strand: any strand: any Count Cut-off: 999 Beginning: -300 End: -300 Window Width: 10The high cut-off value preserves the numbers in the count field which will later be used for peak ranking. On ChIP-cor result page, use the "Enriched Feature Extraction Option" to annotate the ChIP-Seq peak list for colocalization with the qPCR-based binding site list with the following parameters:
From: -250 To: +250 Threshold: 0 Cut-off 999 Switch to Depleted Feature Selection off Ref Feature Oriented off
The resulting annotated peak files are posted below:
NC_000001.9 ChIP_R 110751869 0 17 0 NC_000001.9 ChIP_R 110887050 0 66 1 NC_000001.9 ChIP_R 110960004 0 10 0the R code for making the sensitivity plots is shown below:
# computing the sensitivity curve
table=read.table("GABP_ChIP-Peak_qPCR.sga",as.is=5)
# in case a peaks is near several qPCR peaks (not likely at all)
# sets the count to 1 (acts as a boolean flag)
table[table[,6] > 0, 6] = 1
rank = order(table[,5], decreasing=T)
table=table[rank,]
N=dim(table)[1]; x = 1:N
y = rep(0,N); y[1]=table[1,6]
for(i in 2:N) {y[i]=y[i-1]+table[i,6]}
# making the figure
tot=150 # number of PCR-verified sites: 150 for GABP, 83 for NRSF
par(cex=1.2)
plot(x,100*(y/tot),t="l", col="red", ylim=c(0.0,100), xlim=c(0,20000),
xlab="Peak rank",
ylab="Percent of qPCR true positives found (%)")
legend("bottomright", legend=c("ChIP-Peak(t=10)"),
lty=1, col=c("red"), bty="n")
You can now generate sensitivity plots for other GABP peaks list provided by the
ENCODE consortium in the same way. Keep in mind that in the peak lists provided as
serverr-resident files, the peak ranking score is given in the sixth
rather than fifth field. Once you have computed all curves,
you can superpose them in one Figure. This will tell you how well
ChIP-peak performs as compared to the peak finders that were
used for generating the published peak lists.
Making sensitivity plots based on motif enrichment
You can do this in two alternative ways, one using FindM and the other one using PWMScan. We will present the recipe based on PWMScan.
Open the PWMScan input form. Perform a whole hg18 genome scan using the following parameters:
Genome Assemblies: H.sapiens (March 2006 NCBI36/hg18) Motif Library: JASPAR CORE 2014 vertebrates Motif: MA0062.2 GABPA Bg base composition 0.29, 0.21, 0.21, 0.29 Cut-off P-value: 0.00001 Search Strand: both Non-overlapping matches: checked Reference position 0Note that the authors of the paper use 10-7 as P-value cut-off. This seems to low. However, they used a different program called FIMO to search for PWM matches, and P-values computed by different programs may not be comparable.
Ranking accuracy plots of the kind shown in Figure 6 of the paper can be generated with the following R recipe:
# computing the ranking accuracy curve
table=read.table("NRSFpoly_ChIP-Peak_motifs.sga",as.is=5)
# in case a peaks is near several motif instances (happens)
# sets the count to 1 (acts as a boolean flag)
table[table[,6] > 0, 6] = 1
rank = order(table[,5], decreasing=T)
table=table[rank,]
N=dim(table)[1];
x=seq(50,N-50,50)
y=x; for(i in 1:length(x)) {y[i]=sum(table[1:x[i],6])}
# making the figure
par(cex=1.2)
plot(x,100*y/x,t="l", col="red", ylim=c(0.0,100), xlim=c(0,9000),
xlab="Number of ranked peaks",
ylab="Percent with NRSF motif (%)")
legend("topright", legend=c("ChIP-Peak(t=10)"),
lty=1, col=c("red"), bty="n")