
- PWMTools
- Browse and Download PWMs
- MGA Database
- Other Resources
- References
- Frequently Asked Questions
- What is new
PWMScan - Genome-wide position weight matrix (PWM) scanner
  |
Background |
Alternatively, motifs can be represented as consensus sequences for which both the IUPAC code for nucleotides and the number of allowed mismatches can be specified.
MEME Motif Format
The motif library provided by PWMTools have been originally downloaded from The MEME Suite website. Motifs have then undergone a reformatting process (for more details, please read here).
Matrices from MEME are provided in two formats:
- as letter-probability matrices;
- as integer log-odds weight matrices.
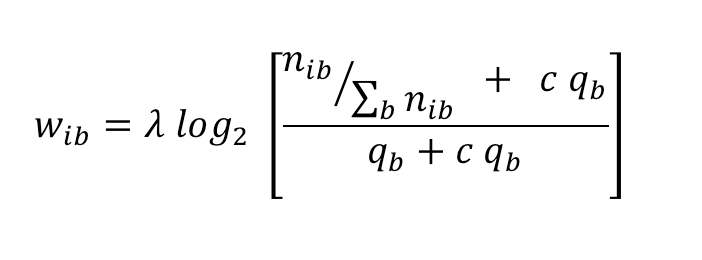
|
(1) |
The relative frequency f'ib of residue b at PWM position i is defined as:
|
|
(2) |
Weights are rounded to nearest integers to allow for efficient computation of the probability distribution for scores expected from random sequences.
For more details, visit the MEME website or our PWMLib site .
|
|
Matrix Formats |
TRANSFAC-style matrices look like this:
AC M00223 XX ID V$STAT_01 XX DT 29.11.1995 (created); ewi. DT 11.03.2003 (updated); dtc. CO Copyright (C), Biobase GmbH. XX NA STATx XX DE signal transducers and activators of transcription XX BF T01575 STAT1alpha; Species: mouse, Mus musculus. BF T01492 STAT1alpha; Species: human, Homo sapiens. BF T01573 STAT1beta; Species: human, Homo sapiens. XX PO A C G T 01 0 0 0 14 T 02 0 0 0 14 T 03 2 12 0 0 C 04 0 12 0 2 C 05 0 9 2 3 C 06 4 2 8 0 G 07 0 0 8 6 K 08 13 1 0 0 A 09 14 0 0 0 A XX BA 14 genomic sequences of 14 different genes XX CC compiled sequences XX RN [1]; RE0003481. RX PUBMED: 7774815. RA Horvath C. M., Wen Z., Darnell jr J. E. RT A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain RL Genes Dev. 9:984-994 (1995). XX
JASPAR-style matrices look like this:
>MA0137.2 STAT1 A [ 208 859 251 10 8 106 23 528 696 53 1900 2030 954 336 417 ] C [1076 496 574 22 14 1921 1900 762 31 30 124 17 263 760 804 ] G [ 415 279 144 11 38 14 7 115 1292 1700 29 23 552 270 425 ] T [ 378 446 1112 2038 2023 44 155 680 66 302 32 15 315 714 431 ]
SSA-style matrices look like this:
TI TATA-box TATA-box FP XX CO 79.00% XX WM -3 -1.02 -0.28 0.00 -1.68 WM -2 -3.05 -2.06 -2.74 0.00 WM -1 0.00 -5.22 -4.38 -2.28 WM 0 -4.61 -3.49 -4.61 0.00 WM 1 0.00 -5.17 -3.77 -2.34 WM 2 0.00 -4.63 -4.73 -0.52 WM 3 0.00 -4.12 -2.65 -3.65 WM 4 0.00 -3.74 -1.50 -0.37 WM 5 -0.01 -1.13 0.00 -1.40 WM 6 -0.94 -0.05 0.00 -0.97 WM 7 -0.54 0.00 -0.09 -1.40 WM 8 -0.48 -0.05 0.00 -0.82 WM 9 -0.48 -0.11 0.00 -0.66 WM 10 -0.74 -0.28 0.00 -0.54 WM 11 -0.62 -0.40 0.00 -0.61 //
PFM-style matrices look like this:
> M00224 STAT1 8.0 18.0 41.9 32.1 23.2 9.7 37.2 29.8 13.9 31.3 19.8 35.2 17.5 37.9 38.1 6.4 59.6 13.5 15.1 11.9 31.2 26.3 9.4 33.2 0 0 0 100 0 0 0 100 3.9 90.9 2 3.2 0 100 0 0 0 32.5 67.5 0 0 0 100 0 3.8 0 94.9 1.2 100 0 0 0 100 0 0 0 36.3 8.8 29.5 25.5 11.1 11.1 11.1 66.7 19.8 10.5 69.6 0 24.8 12.7 46.6 15.9 12.5 36.7 49.2 1.5 31.8 12.5 39.7 16
LPM-style matrices look like this:
> letter-probability matrix V_STAT1_01: alength= 4 w= 21 nsites= 100.1 E= 0 0.080000 0.180000 0.419000 0.321000 0.232232 0.097097 0.372372 0.298298 0.138723 0.312375 0.197605 0.351297 0.175175 0.379379 0.381381 0.064064 0.595405 0.134865 0.150849 0.118881 0.311688 0.262737 0.093906 0.331668 0.000000 0.000000 0.000000 1.000000 0.000000 0.000000 0.000000 1.000000 0.039000 0.909000 0.020000 0.032000 0.000000 1.000000 0.000000 0.000000 0.000000 0.325000 0.675000 0.000000 0.000000 0.000000 1.000000 0.000000 0.038038 0.000000 0.949950 0.012012 1.000000 0.000000 0.000000 0.000000 1.000000 0.000000 0.000000 0.000000 0.362637 0.087912 0.294705 0.254745 0.111000 0.111000 0.111000 0.667000 0.198198 0.105105 0.696697 0.000000 0.248000 0.127000 0.466000 0.159000 0.125125 0.367367 0.492492 0.015015 0.317682 0.125874 0.396603 0.159840
PWM-style matrices look like this:
>MA0137.2 STAT1
-132 105 -32 -46
72 -7 -90 -22
-105 14 -185 110
-570 -456 -556 197
-602 -522 -378 196
-230 188 -522 -357
-450 187 -622 -175
2 55 -218 38
42 -407 131 -298
-330 -412 171 -79
187 -207 -417 -403
196 -494 -450 -512
87 -99 8 -73
-63 55 -95 46
-32 63 -29 -27
|
>CTCF-motif -0.96 -0.89 0.00 -0.72 -0.59 -1.83 0.00 -1.22 -2.64 0.00 -3.50 -3.10 -4.48 0.00 -4.70 -4.71 0.00 -2.54 -2.29 -1.84 -2.12 -0.12 0.00 -3.09 -0.31 0.00 -2.13 -0.32 0.00 -3.88 -2.90 -2.74 -4.15 -4.69 0.00 -5.28 0.00 -4.05 -0.05 -3.56 -3.20 -4.33 0.00 -0.89 -4.02 -5.28 0.00 -4.95 -3.36 -2.68 0.00 -2.74 -1.83 0.00 -2.99 -2.21 0.00 -2.37 -0.04 -2.77 -1.73 0.00 -0.53 -1.17 -0.96 -0.08 -1.95 0.00 0.00 -0.74 -0.54 -1.63
|
|
|
Method Overview |
The Position Weight Matrix (PWM) is the most commonly used tool to describe the DNA binding motif of a transcription factor. A PWM contains weights for each base at each motif position. A PWM score can be computed for any base sequence of the same length by simply summing up the corresponding weights from the PWM.
PWMscan is able to:
- Scan a PWM against a genome
- Compute the p-value corresponding to a threshold score (or cut-off), or the score corresponding to a p-value
PWMscan has the following characteristics:
- Menu-driven access to genomes of more than 20 model organisms
- Access to large colllections of PWMs from MEME and other databases
- PWMs are supplied by copy&paste or file upload
- Support of various PWM formats: JASPAR, TRANSFAC, plain text, etc.
- Cut-off values defined as PWM-scores, match percentage, or P-values
- Automatic conversion of base-probabilities and to log-odds scores
- Output provided in various formats: BEDdetail, SGA, FPS, etc.
- Direct link to UCSC genome browser for visualization of results
- Action buttons to transfer match list to downstream analysis tools (ChIP-Seq and motif analysis tools)
The program takes as input a Position Weight Matrix, the background probabilities for the letters of the DNA alphabet and a threshold score or a p-value. The search is carried out across the entire genome sequence. It uses PWMs, such as those available in the Transfac or Jaspar databases, as well as plain-text PWMs. It computes all occurrences of the matrix (or matches) in the genome sequence for a given p-value threshold or score. The threshold score can also be expressed as percentage score from 0% (e.g. minimum score) to 100% (e.g. maximum score).
The algorithm is very fast and allows for large-scale analysis. The method is based on two alternative search sequence engines:
- Bowtie, a fast memory-efficient short read aligner using indexed genomes (see References below);
- matrix_scan, a C program implementing a conventional search algorithm that has been optimized for rapid score computation and drop-off strategy.
The match list is provided in various genome annotation formats, including BEDdetail, FPS, and SGA. BEDdetail is an extension of BED format that is used to enhance the track display page. For PWMScan, we use BEDdetail format to include the name of the PWM as well as the p-value associated to the motifs identified by PWMScan.
PWMScan is meant to support ChIP-seq data analysis and is designed to be interoperable with other tools from our group, in particular tools from the ChIP-Seq and SSA servers.
List of arguments for PWMs
Mandatory Input arguments are:- Position Weight Matrix (text format or file to upload)
- Cut-off or threshold score (aboslute value, percentage, or p-value)
- background probabilities
- genome assembly (DNA sequence to scan)
- Search Strand (both or single strand on genome)
- Offset (match position relative to the motif sequence)
- Non-overlapping matches (filter out overlapping matches, and only retain the match with the highest score)
- Lowest Log-odds Score (for JASPAR/TRANSFAC conversion; default=-10000)
- Pseudo-count fraction (for PFM/LPM conversion; default=10-6)
- Log-odds Scaling Factor (default=100)
List of arguments for Consensus Sequences
Mandatory Input arguments are:- Consensus sequences
- Allowed Mismatches (default=0)
- Search Strand (both or single strand on genome)
- Offset (match position relative to the motif sequence)
- Non-overlapping matches (filter out overlapping matches, and only retain the match with the highest score)
References
-
Ambrosini G., Groux R., and Bucher P.
PWMScan: a fast tool for scanning entire genomes with a position-specific weight matrix
Bioinformatics, Bioinformatics 2018; 34(14):2483-2484 -
Iseli C., Ambrosini G., Bucher P., and Jongeneel CV.
PMID:17593978
Indexing Strategies for Rapid Searches of Short Words in Genome Sequences
PLoS ONE. 2007; 2(6):e579 -
Langmead B., Trapnell C., Pop M., and Salzberg SL.
PMID:19261174
Ultrafast and memory-effcient alignment of short DNA sequences to the human genome.
Genome Biol. 2009; 10(3):R25
The STAT1 PWM is the one from the JASPAR database (ID MA0137.3) and is provided as a custom matrix in integer log-odds format.
The NANONG PWM is the one from the HOCOMOCO database (ID NANOG_MOUSE.H11MO.1.A) and is selected from the HOCOMOCO v11 (FULL) library. Transcription factor binding predictions are provided for regions that are enriched in the H3K27me3 histone mark in ES cells. These regions are defined in a user-provided BED file (Mouse_mm9_ES.H3K27me3-mikkelsen2007.bed)
