ChIP-Seq Technical Document
1. Data representation and file formats
The ChIP-Seq programs use SGA (Simple Genome Annotation) files as INPUT and OUTPUT. SGA files are used to represent ChIP-Seq data as well as other genome annotations such as the location of TSSs or matches to consensus sequences.
SGA is a single-line-oriented and tab-delimited format with the following five
required fields:
- Sequence name (Char String)
- Feature (Char String)
- Sequence Position (Integer)
- Strand (+/- or 0)
- Tag Counts (Integer)
Any number of additional fields may be added containing application-specific information.
Here are some example records:
|
Chip-seq programs require SGA files to be sorted by chromosome name, position and strand.
As a rule of thumb, use the following UNIX command to sort SGA files:
setenv LANG C; sort -s -k1,1 -k3,3n -k4,4
or in Bash environment:export LANG=C; sort -s -k1,1 -k3,3n -k4,4
Tag counts for a particular chromosome/position/strand combination may be distributed over several lines. For instance:NC_000001.9 stim 559778 + 2Could be represented by two lines:
NC_000001.9 stim 559778 + 1 NC_000001.9 stim 559778 + 1
SGA is a generic format that can be used for representation of other experimental data as well as genome annotations. The ChIP-Seq server offers a rich collection of public data sets from ladmark experiments, which have been widely used by the bioinformatics community for benchmarking and testing new algorithms. These collections include mass genome annotation data such as CAGE and DBTSS, or even non-experimental annotations such as cross-genome conservation scores.
The SGA format differs in one very important aspect from similar formats such as BED or GFF. It is required to be sorted by genome positions. The fact that ChIP-Seq programs operate on sorted input files enables them to generate results in one sweep through the genome. We support data upload in SGA, FPS, BED, GFF and BAM formats. FPS is the specific format used by the Signal Search Analysis server, a motif discovery platform developed by our group. Both the ChIP-Seq and Signal Search Analysis servers use RefSeq IDs including version numbers for internal representation (Figure 1).
In a data analysis pipeline, the SGA file is typically generated from a variety of richer formats, such as the Solexa genome mapping files, BED (Browser Extensible Data) files, GFF (Generic Feature Format) or BAM (Binary Alignment/Map) files.
It is also interconvertible with the FPS (Functional Position Set) format used by our Signal Search Analysis programs (SSA).
SGA may also be converted on output into other formats, in particular BED or WIG (Wiggle Track Format) formats for data visualization, and FPS for sequence extraction.
We will describe the FPS format herein. We will first focus on GFF and BED/WIG conversion rules.
1.1 SGA fields
SGA files typically represent a list of sequences reads from ChIP-Seq experiments that have been mapped to a reference genome.
Column 1 is the sequence identifier or the chromosome access number. In all server-resident SGA files we use versioned 'NC_' accession numbers which identify primary entries in NCBI's nucleotide databases.
This is not a requirement. You upload sga files with chromosome names as identifiers. However, you will not be able to jointly analyze your data with the server-resident files. Moreover, our programs for sequence retrieval and sequence analysis only recognize 'NC_' or 'NT_' reference identifiers as well as some EMBL/Genbank identifiers.
Column 2 is the feature name of the genomic feature currently represented by the SGA file. Feature names may contain any characters, but must avoid tabs and whitespaces.
Chip-Seq data from different experiments can be merged into one SGA file, using the feature name as an identifier for the source experiment.
Column 3 is the start coordinate of the feature (or tag sequence), in 1-based integer coordnates.
Colum 4 is the strand of the feature: + for positive strand (relative to the landmark), - for minus strand, and 0 for features that are not stranded.
Colum 5 is the count of the feature, an integer number indicating the number of sequence tags that have been mapped to the current position. The position always corresponds to the first base (5'end) of the sequence tag
regardless of the orientation.
Additional fields can be used for different purposes. It could be a list of feature attributes in the format tag=value as well as comments. Spaces are allowed in this field as well as tabs, whereas spaces are not allowed in the feature field.
Sequence regions can be defined by pairs of lines with the same feature name but opposite orientations ('+' followed by '-').
1.2 GFF and BED conversion rules
For SGA to GFF conversion we comply with version 3 format (GFF3).
GFF3 format is a flat tab-delimited file. The first line of the file is a comment that identifies the file format and version. This is followed by a series of data lines, each one of which corresponds to an annotation. Here is a miniature GFF3 file:
##gff-version 3 ctg123 . exon 1300 1500 . + . ID=exon00001 ctg123 . exon 1050 1500 . + . ID=exon00002 ctg123 . exon 3000 3902 . + . ID=exon00003 ctg123 . exon 5000 5500 . + . ID=exon00004 ctg123 . exon 7000 9000 . + . ID=exon00005
The ##gff-version 3 line is required and must be the first line of the file. For a detailed description of the 9 columns annotation please refer to GFF3 format.
The sample SGA lines above will be converted as follows:
##gff-version 3 chr1 ChIPSeq stim 559139 559139 1 - . 1 chr1 ChIPSeq stim 559333 559333 1 + . 2 chr1 ChIPSeq stim 559356 559356 1 - . 3 chr1 ChIPSeq stim 559765 559765 1 - . 4 chr1 ChIPSeq stim 559766 559766 3 + . 5 chr1 ChIPSeq stim 559767 559767 1 + . 6 chr1 ChIPSeq stim 559768 559768 1 + . 7 chr1 ChIPSeq stim 559777 559777 3 + . 8 chr1 ChIPSeq stim 559778 559778 2 + . 9 ...The Web Server provides the choice of uploading GFF files for ChIP-Seq analysis. In such case, a control on the GFF syntax is performed, and the format must comply to version 3. Therefore the format is a flat tab-delimited file with the following requirements:
- we should have at least 8 colums
- colum 1 is the ID of the landmark used to establish the coordinate system for the current feature. For correct SGA conversion the ID should be of type chr# or NC_* , even though other sequence IDs are accepeted
- colum 2 is the source which is a free text qualifier intended to describe the algorithm or experiment that generated this feature. It is not necessary to specify a source. If there is no source, put a "." (a period) in this field
- colum 3 must report the feature type (or name); this field is required
- colums 4 and 5 are the start and end of the feature, in 1-based integer coordinates, relative to the landmark given in column 1. Start is always less than or equal to end
- colum 6 must be an integer value reporting the feature score (or count). This field is required
- colum 7 must be the feature strand (+, -, . or ? are allowed values)
- colum 8 is the feature phase. For features of type "CDS", the phase indicates where the feature begins with reference to the reading frame. If there is no phase, put a "." (a period) in this field.
The columns not mentioned above are ignored. If field 6, the feature score, is not a positive integer the SGA conversion fails.
If sequences are specified as UCSC chromosome identifiers, they will be converted into NC identifiers if a genome assembly is specified.
Here's an example of a GFF-based track taken from the UCSC browser:
chr22 TeleGene enhancer 1000000 1001000 500 + . touch1 chr22 TeleGene promoter 1010000 1010100 900 + . touch1 chr22 TeleGene promoter 1020000 1020000 800 - . touch2
Specifying H.sapiens (March 2006) genome assembly, the UCSC tranks above will be converted into the following SGA lines:
NC_000022.9 enhancer 1000000 + 500 NC_000022.9 promoter 1010000 + 900 NC_000022.9 promoter 1020000 - 800
The Web Server also supports data upload in BED format, and SGA output might be converted into WIG or BED format that can be easily linked to the UCSC genome browser displaying information about the genomic environment of the corresponding feature.
BED lines have three required fields and nine additional optional fields. The number of fields per line must be consistent throughout any single set of data in an annotation track.
The first three required BED fields are:
1. chrom - The name of the chromosome (e.g. chr3, chrY, chr2_random) or scaffold (e.g. scaffold10671).
2. chromStart - The starting position of the feature in the chromosome or scaffold. The first base in a chromosome is numbered 0.
3. chromEnd - The ending position of the feature in the chromosome or scaffold. The chromEnd base is not included in the display of the feature. For example, the first 100 bases of a chromosome are defined as chromStart=0, chromEnd=100
(position 0 to 99 on zero-based numbering system, 1-100 on a standard EMBL/GenBank/GFF/SGA/FPS numbering system).
In addition, we define the strand on the 6th field of the BED line ('0' is converted into '+' on output).
On input, we check whether the strand field is defined or not. If so, we record the 5'end position of each line for generating the correspondign SGA file. If the strand field is missing, we assume that the BED file stores peak positions and we record the center position of each line.
To convert BED format to SGA format, users should pay attention to the fact that the start coordinate of a feature in BED is a 0-based integer number, namely the first base in a chromosome is numbered zero, whereas the start and end of a feature in SGA is in 1-based integer coordinates. Therefore when converting BED to SGA the start coordinate should be incremented by 1.
Here is an example of an annotation track in BED format that is generated by the partitioning tool (ChIP-Part) on the Web:
browser position chr1:556608-1556608 browser full refGene track name=robertson07_22846 description="H_sapiens/STAT1-stim Partitioning" visibility=1 color=200,100,0 chr1 556607 556831 + chr1 559764 559815 + chr1 863348 863972 + ....
When more appropriate, we provide Wiggle Track format (WIG). The WIG format is for display of dense, continuous data such as ChIP-Seq peaks.
Wiggle format is line-oriented. For wiggle custom tracks, the first line must be a track definition line, which designates the track as a wiggle track and adds a number of options for controlling the default display.
Wiggle format is composed of declaration lines and data lines. There are two options for formatting wiggle data: variableStep and fixedStep. These formats were developed to allow the file to be written as compactly as possible.
For ChIP-peak we use variableStep that is for data with irregular intervals between new data points and is the more commonly used wiggle format. It begins with a declaration line and is followed by two columns containing chromosome positions and data values:
variableStep chrom=chrN [span=windowSize] chromStartA dataValueA chromStartB dataValueB ... etc ... ... etc ...
For example, this variableStep specification is used by ChIP-peak on the Web:
browser position chr1:0-1000000 browser full refGene track type=wiggle_0 name="robertson07_stim" description="STAT1-stim" visibility=full autoScale=off variableStep chrom=chr1 span=150 1060827 102 2311846 120 6217246 167
1.3 FPS format
The FPS format is used by the SSA programs. These programs typically do not use sequences as input, but lists of computer-readable pointers to sequence positions in a database. Such a list of pointers is called a Functional Position Set, or FPS. Each pointer contains a sequence id, a position, and two flags, one indicating the strand (+ or -), the other one the topology (1=linear, 0=circular). The Eukaryotic Promoter Database (EPD) is an example of a functional position set.
Here is an example of FPS lines for EPD:
FP Pv snRNA U1 :+S EM:J03563.1 1+ 352; 17001.098 FP Ath snRNA U2.5 :+S EM:AL353994.1 1- 73709; 24016.116 FP Ath snRNA U5 :+S EM:X13012.1 1+ 678; 23040. FP Ta histone H3 :+S EM:X00937.1 1+ 186; 07001. FP Ta histone H4 :+S EM:X00043.1 1+ 669; 07002. FP Ath EF-1a' A1 :+S EM:U63815.1 1+ 39357; 35037.
FPS is an old fashion fixed-length field-width format (see the EPD User Manual).
The four crucial elements are the sequence id (column 4), the topology (column5), the orientation (column6), and the position (column 7). SSA recognizes only 'NC_' or 'NT_' numbers and some EMBL/Genbank identifiers. The sequence identifier (column 4) is composed of two parts: a source ID (EM for Genbank/EMBL, EU for RefSeq genome sequence identifiers), and the accession number.
1.4 BAM format
The BAM (Binary Alignment/Map) format is derived from the SAM format which is a generic alignment format for storing read alignments against reference sequences.
BAM to SGA conversion is carried out using the BEDTools utilities which provide a useful set of tools for comparing genomic features based on four widely-used formats: BED, GFF/GTF, VCF, and SAM/BAM. Among the supported utilities, we find the bamToBed program for converting BAM alignments to BED format.
Even though BAM is a rather compact format, for most applications, the SGA format represents the best trade-off between costs in computer resources (i.e. disk space and data processing time) and data quality.
2. Input Data on the Web Server
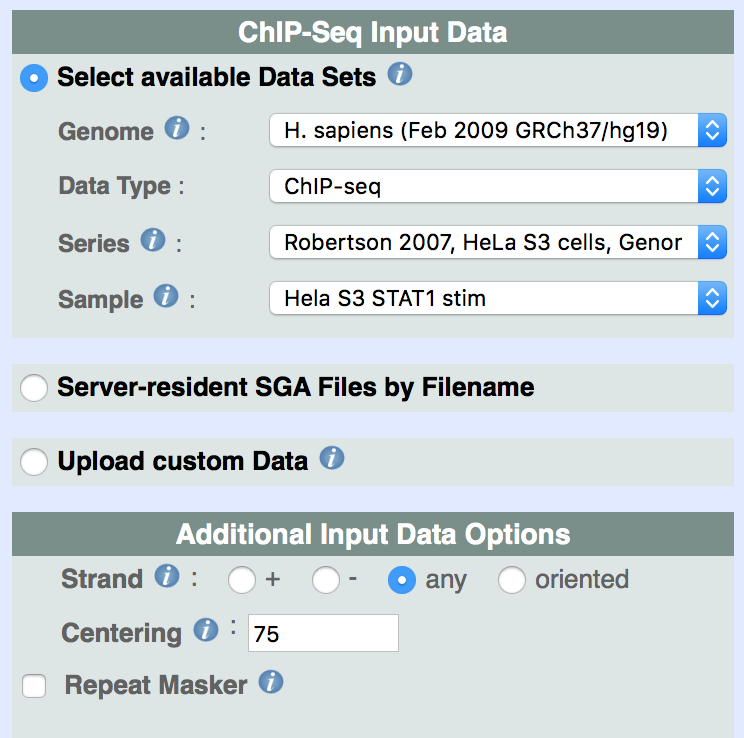
As previously stated, all ChIP-Seq tools use use sga files as inputs and outputs.The user interface has three main input modules for selecting data (see Fig. 1):
-
i. Select available data sets from the Mass Genome Annotation (MGA) Data Repository;
ii. Server-resident SGA files only relevant for local users;
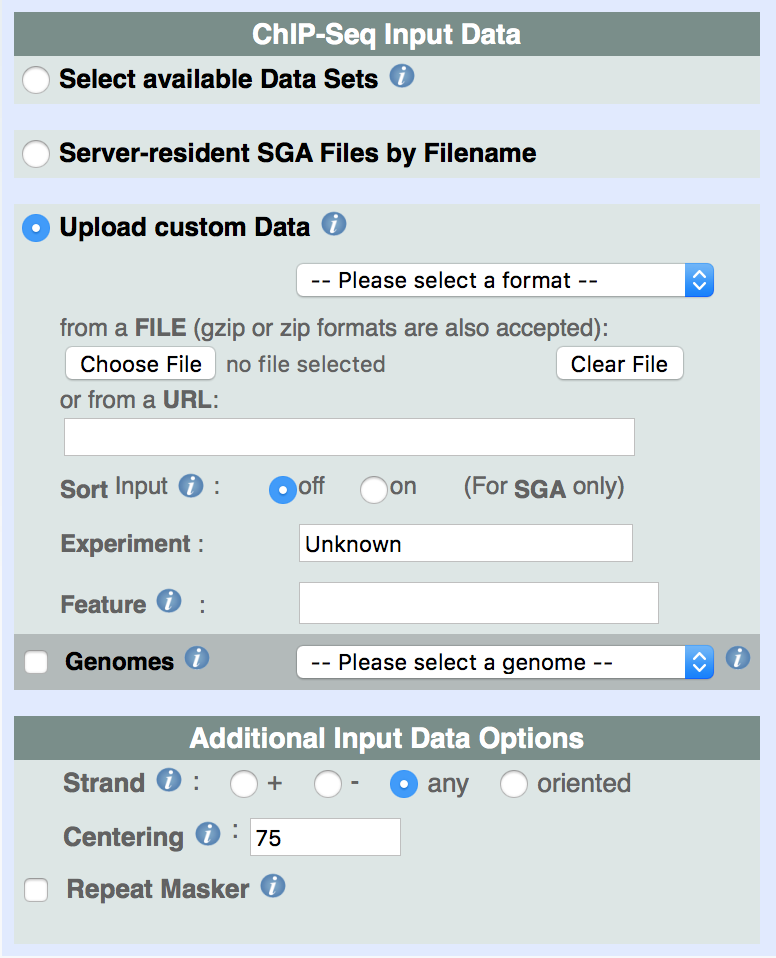
iii. Upload of custom ChIP-Seq data (in SGA, BED, GFF, BAM or FPS format).
|
|
Available Data Sets from the MGA Data Repository include published next generation sequencing (ChIP-seq, DNAse-seq, MNAse-seq, etc.) data sets that have become available starting from the year 2007, as well as other genome annotation (such as gene start sites, SNPs, etc.) and sequence-derived data (such as Phylop Conservation Score). You will find a few examples below:
- Barski et al. (2007): human CD4+ cell lines Histone modifications, POL II, CTCF (~2 millions tags per experiment)
- Mikkelsen et al. (2007): four mouse cell lines Histone modifications (~2 millions tags per experiment)
- Robertson et al. (2007). INF-gamma stimulated HeLa cells STAT1 (>20 million tags per experiments)
- Boyle et al. (2008): CD4+ cell lines Open chromatin studies (~10 millions tags per experiment)
- Wang et al. (2008): human CD4+ cells lines Histone acetylations and methylations (3,4 millions tags per experiments)
- Schones et al. (2008): human CD4+ cells lines Regulation of nucleosome positioning (100 millions tags per experiments)
- CAGE, ENSEMBL, UCSC and EPD TSS collections
- SwissRegulon annotations of regulatory sites
- ENSEMBL POLYA
- SNP collections from 1000 Genomes Project, dbSNPs, etc.
- Phylop base-wise Conservation Score tracks from UCSC
- Phastcons tracks from UCSC
- Lists of motif matches from public Motif Databases (JASPAR, HOCOMOCO, etc.)
By selecting the genome assembly, three additional pull-down menus will allow the selection of the ChIP-Seq experiment, the type of feature, and the chromosomal region to be studied (by default the analysis span across the whole DNA).
Server-resident SGA files by Filename are server-resident files which are only relevant for users at SIB or EPFL. Users have the option of specifying the feature name or type (which is the SGA second field) and the ChIP-Seq experiment. These input fields can be left blank, in which case all input tag reads are processed.
Upload of custom input files is also permitted. In such case, three formats are allowed, provided they comply with the rules previouly described: SGA, GFF, and FPS. As in the case of server-resident files for local users, one can optionally specify the feature name and the experiment from which data was produced. If no feature is given then all input tags are processed. When the data file contains more than one feature, in case we want to analyse one specific feature, we need to input the feature name. The experiment field is only used as a title in the output, it has no relation to the input data file.
Additional Input Data Options, common to all type of data, are provided in order to select the strand of the feature ('+', '-', or both strands), the centering of all input tag positions to estimated center positions of DNA fragments, and the filtering of repeat loci (Repeat Masker) within the input data file. Centering might be useful in some cases for correlation analysis or peak detection (see the Tutorial example). The Repeat Masker option removes all tags that fall into repeats that are annotated as such in the USCS browser database. If repeat masking is used for uploaded files, the Genomes checkbox need to be checked, and a corresponding genome assembly needs to be specified.
Server-resident SGA files are sorted by sequence ID (or chromosome name), position and strand according to the following rule:
setenv LANG C; sort -s -k1,1 -k3,3n -k4,4
or in bash environment:
export LANG=C; sort -s -k1,1 -k3,3n -k4,4
When uploading a custom SGA file, the back-end programs perform a sorting order check and
in case of failure, the programs still perform the sorting, but they return a warning message asking the user to activate the Sort checkbox in the Input Form:
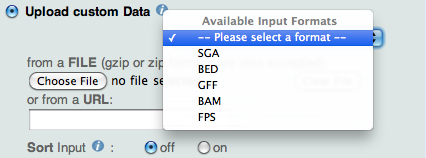
|
| Figure 2. ChIP-Seq Upload custum Data form : Available Formats and Sort checkbox |
In case the custom file is BED, GFF, BAM or FPS, the Web interface first converts the file into SGA and then sorts it according to the above rule.
For chromosome name to RefSeq ID conversion, sequence extraction, comparison with server-resident public data, or data visualization at the UCSC genome browser, the user is required to specify the species assembly of the uploaded data by means of a Genomes menu button as shown here below:

|
| Figure 3. ChIP-Seq Genomes checkbox |
3. Analysis Parameters
Common to all ChIP-Seq analysis programs is the input data file: a sorted list of sequence tag reads in SGA format. ChIP-Seq programs accept two types of parameters: general and program-specific parameters.
General parameters are the feature name and strand as well as a cut-off value for the feature count or score.
The feature parameter is a name that is given to the ChIP-seq tags in the SGA file and it is generally related to the corresponding ChIP-Seq experiment. If no feature is given then all input tags are processed. The strand is used to restrict the analysis to either the positiive or negative strand or both strands. If no strand is specified, features on both strands are processed.
A count cut-off value can be optionally specified to reset scores above the cut-off threshold to the cut-off. This is useful when we want, for instance, to count all tag reads mapping to the same position and strand only once.
In what follows we will describe Program-specific parameters.
3.1 ChIP-Cor parameters
The program reads a ChIP-seq data file in SGA format and generates a tag correlation histograms showing the abundance of the 'target' feature as a function of the distance from the 'reference' feature.
ChIP-Cor parameters are the following:
- Range (From-To): it is the relative distance (in bp) between the two features that are being correlated;
- Window width: it defines the histogram step size or bin (in bp);
- Histogram normalization:
- Raw counts: histogram bin entries display the product between reference and target tag counts at the corresponding relative distance;
- Count Density: histogram entries are normalized by the total number of reference tag counts and the window width. The measurement is therefore a 'count density per base-pair' of the target feature relative to the reference feature.
- Global normalization: histograms entries are normalized by the total number of reference and target counts and the window width. It shows a count fold-change of the target feature relative to the reference feature (local target feature frequency over global mean target feature frequency).
On ChIP-Cor the strand feature presents the oriented option. Requiring oriented strand processing means taking into account the orientation of the reference tags and therefore reverting the chromosome axis each time the reference feature is on the negative strand.
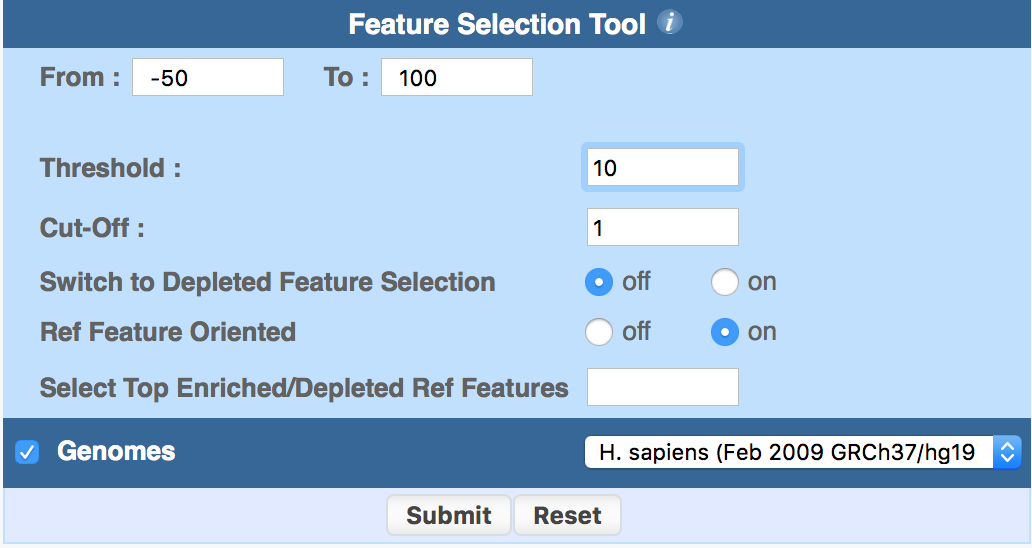
3.2 ChIP-Score (Feature Selection)
ChIP-score is offered as a post-processing feature by the output of ChIP-Core (Feature Selection Tool).
Similarly to ChIP-Cor, the program reads ChIP-seq data files in SGA format and compares two genomic features, a reference and a target feature, with respect to their relative position, and selects those reference features that are enriched in target feature counts within a given range according to a given (count) threshold or score.
By default, the program extracts feature A tags that are enriched in feature B tags. If the Switch to Depleted Feature Selection radio-button is selected, the program extracts sites that are depleted in feature B.
ChIP-Score parameters are the following:
- Range (From-To): it is the relative distance (in bp) between the two features that are being correlated;
- Threshold: it selects those reference positions which:
- have cumulative target tag count ≥ Threshold
- or have cumulative target tag count < Threshold
3.3 ChIP-Peak parameters
The program reads a ChIP-seq data file in SGA format and detects signal peaks for ChIP-tag positions. Tag counts are generally centered to an estimated center-position of DNA fragments before using the peak recognition program. ChIP-Peak implements a rather simple method that works as follow. The number of tags is counted in a sliding window of fixed width w. Performance is enhanced by considering only those windows which are centered around tag positions. At the end, all windows which have a cumulative tag number greater or equal to a threshold value, and in addition are locally maxima within a so-called 'vicinity range', are reported as peaks.
ChIP-Peak parameters are the following:
- Window width: it defines the integration range (in bp) of tag counts
- Vicinity range: it defines the minimal distance (in bp) between reported peaks. Only peaks that have maximal tag counts within the vicinity range are selected
- Peak Threshold: it selects as peaks those positions which:
- have cumulative tag count ≥ Threshold
By activating the Refine peak position checkbox, the program will execute a
post-processing stage that recomputes each peak position by weighting each of the
tag read positions within the given integration (window) range by its tag counts.
It's a weighted mean of tag positions within 'window width':
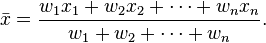
where wn indicates the tag counts at posintion xn.
On the Web, the Peak Threshold parameter can be expressed as both minimal tag counts required for a signal (Tag counts input box) and relative enrichment factor compared to the average background signal (Relative enrichment factor input box). By default, we set both tag count and fold enrichment factor thresholds to 10. The highest threshold in terms of tag count is then selected by the tool.
3.4 ChIP-Part parameters
The program reads a ChIP-seq data file in SGA format and heuristically partitions the data by optimizing a function that scores the partitioning into signal-rich and poor regions of the input read-count (or tag-count) distribution. It implements, in essense, a broad peak caller algorithm used for broad regions of enrichment found in ChIP-seq experiments such as those targeted at histone marks. ChIP-Part outputs a list of signal-enriched regions in SGA format in two lines (beginning, and end coordinates of regions).
ChIP-Part parameters are used to optimize a partitioning scoring function by means of a fast dynamic programming algorithm:
- Count Density threshold
- Transition penalty
- Transition penalties
- Signal-rich region scores: length x (local count-density - threshold)
- Signal-poor region scores: length x (threshold - local count-density)
On the Web, the Threshold parameter can be expressed as both average background count density (Count density box) and relative enrichment factor compared to an average background density (Relative enrichment factor input box). The highest threshold in terms of tag count density is then selected by the tool. A region must have a count density higher than the count density threshold in order to be considered as a signal-rich DNA stretch.
3.5 ChIP-Center parameters
The program reads a ChIP-seq data file in SGA format, and moves ChIP-tag positions corresponding to a given feature to estimated center-positions of DNA fragments. The output is a list of centered un-oriented tag counts.
ChIP-Center has a unique parameter:
- Tag shift: defined as the shift amount for centering tag positions
4. Output Data on the Web Server
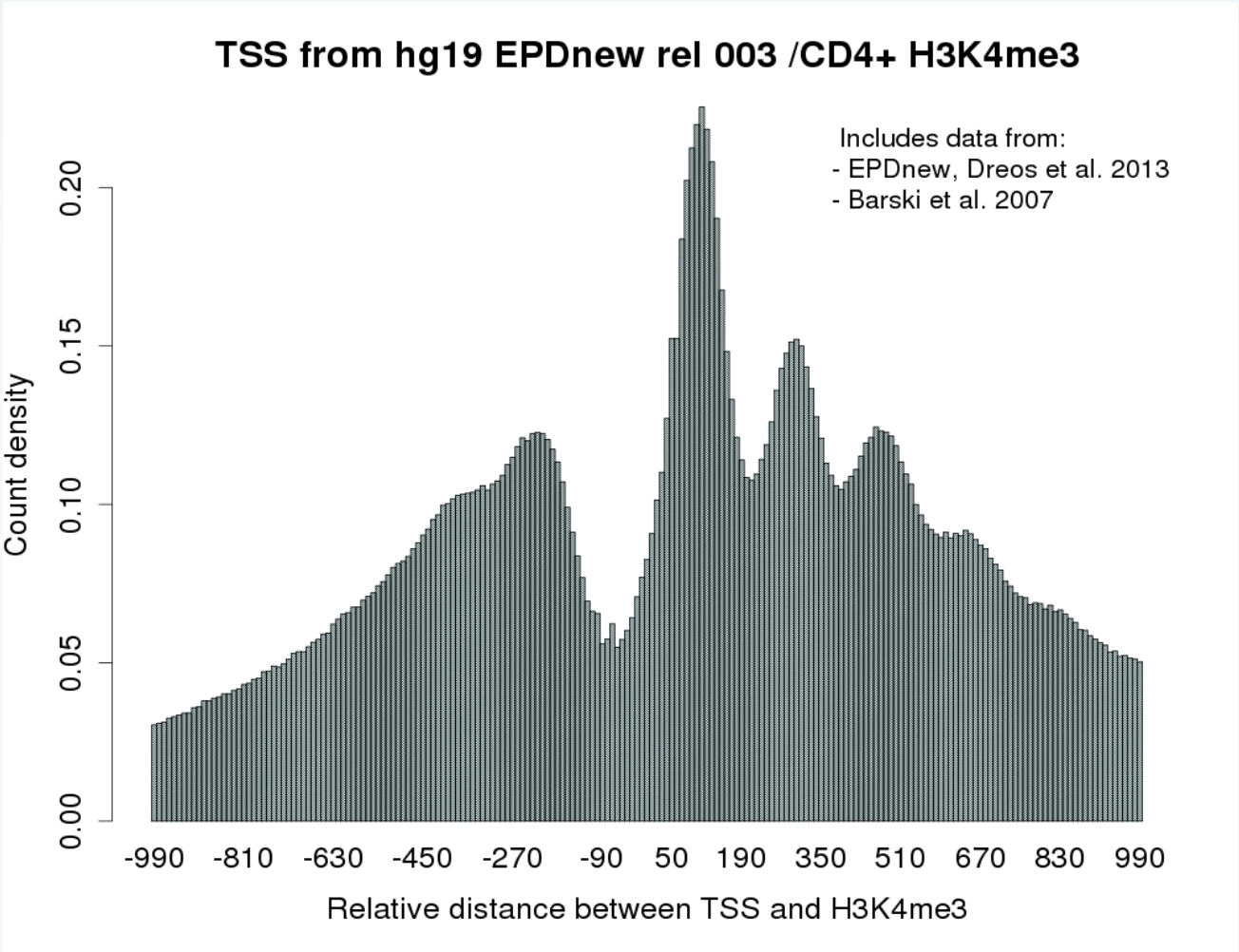
All ChIP-Seq programs, except from ChIP-Cor, generate results in SGA format. ChIP-Cor outputs an aggregation plot (AP) or feature correlation histogram which is a text file indicating the frequency of the target feature as a function of its relative distance to the reference feature.
The Web server provides many more features for displaying results, data comparison with other sources, as well as links and navigation buttons to both external and internal resources.
ChIP-Cor on the Web provides:
- A count correlation histogram graph (in addition to the text file)
- A feature extraction option that allow the selection of reference features that are either enriched or depleted in the target feature. This task is performed by the ChIP-Score application.
|
|
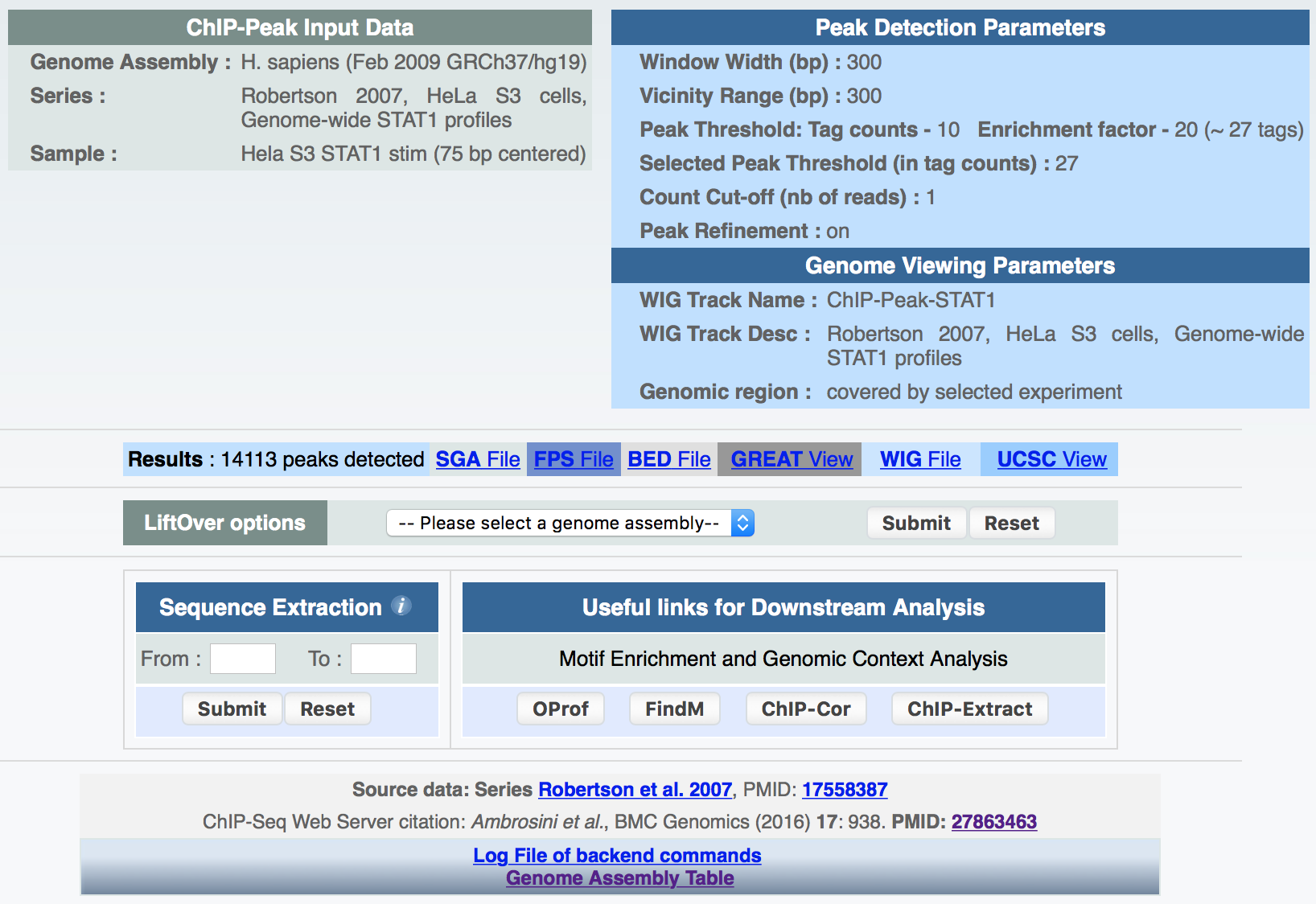
ChIP-Peak on the Web provides, whenever possible, the following information:
- A list of peak positions in other formats besides SGA such as BED, FPS and WIG together with a hyperlink to the UCSC genome browser for data visualization within a broader genomic context;
- A link to the GREAT tool for functional annotations of peak regions;
- Lift over of peak files to different genome assemblies;
- Download of sequences around peak center positions by means of a sequence extraction tool;
- Direct links to other resources/tools developed by our group (SSA and ChIP-Seq) for motif and genomic context analysis.
|
ChIP-Part on the Web provides, in addition to generating a two-line-oriented SGA file representing signal-reach DNA regions, the following information:
- Other data formats such as GFF, FPS, and BED as well as a hyperlink to the UCSC genome browser for rapid comparison with genome annotations;
- Some statistics about the results of the partitioning algorithm;
- Lift over of data files to different genome assemblies;
- Download of sequences around transition boundaries (5' depleted/3' enriched) by means of a sequence extraction tool;
- Direct links to other resources/tools developed by our group (SSA and ChIP-Seq) for motif and genomic context analysis.
|
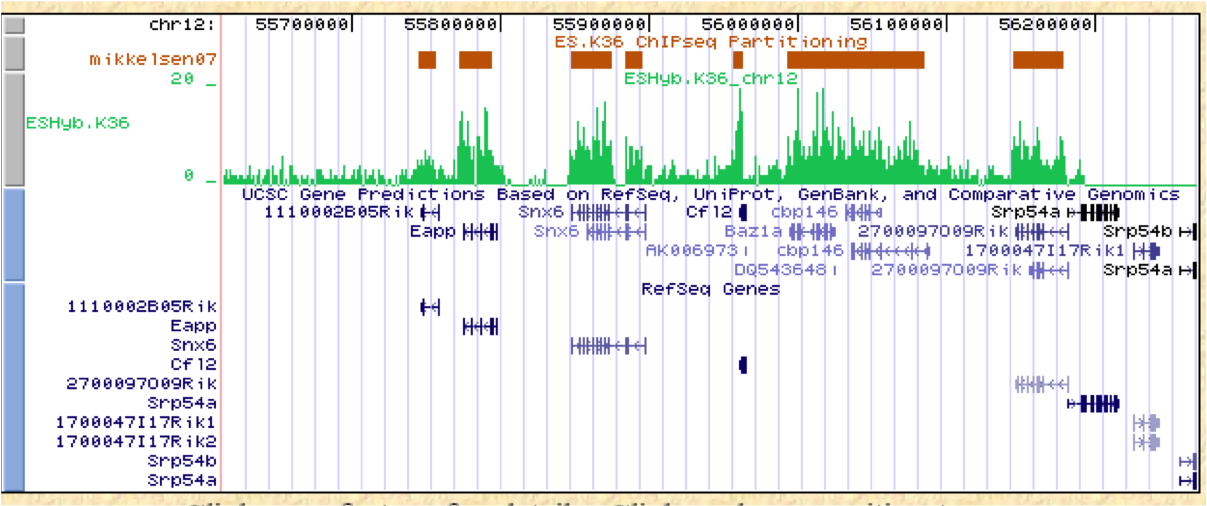
|
Figure 6. Mikkelsen07: results of ChIP-part program (ESHyb.K36 BED file - UCSC custom track) ESHyb.K36: WIG file from Mikkelsen et al. (Genome-wide maps of chromatin state in pluripotent and lineage-committed cells) |
