EPD tutorial
Background.
This tutorial describes how to generate a
validated promoter collection (represented as transcription start
sites, TSS), check its quality and select promoters from it. In
section 1 you will learn how to experimentally validate a promoter
collection downloaded from public repositories. You will
use CAGE
data available on our server as the experimental evidence. The
flowchart underlying this section is presented in the Figure
1. Section 2 will be focused on describing quality checks that can
be applied to promoters lists that rely on studying
promoter-specific feature such as distribution of core promoter
elements or histone marks around TSS. Finally, section 3 will
focus on how to select promoters based on their expression in
specific samples.
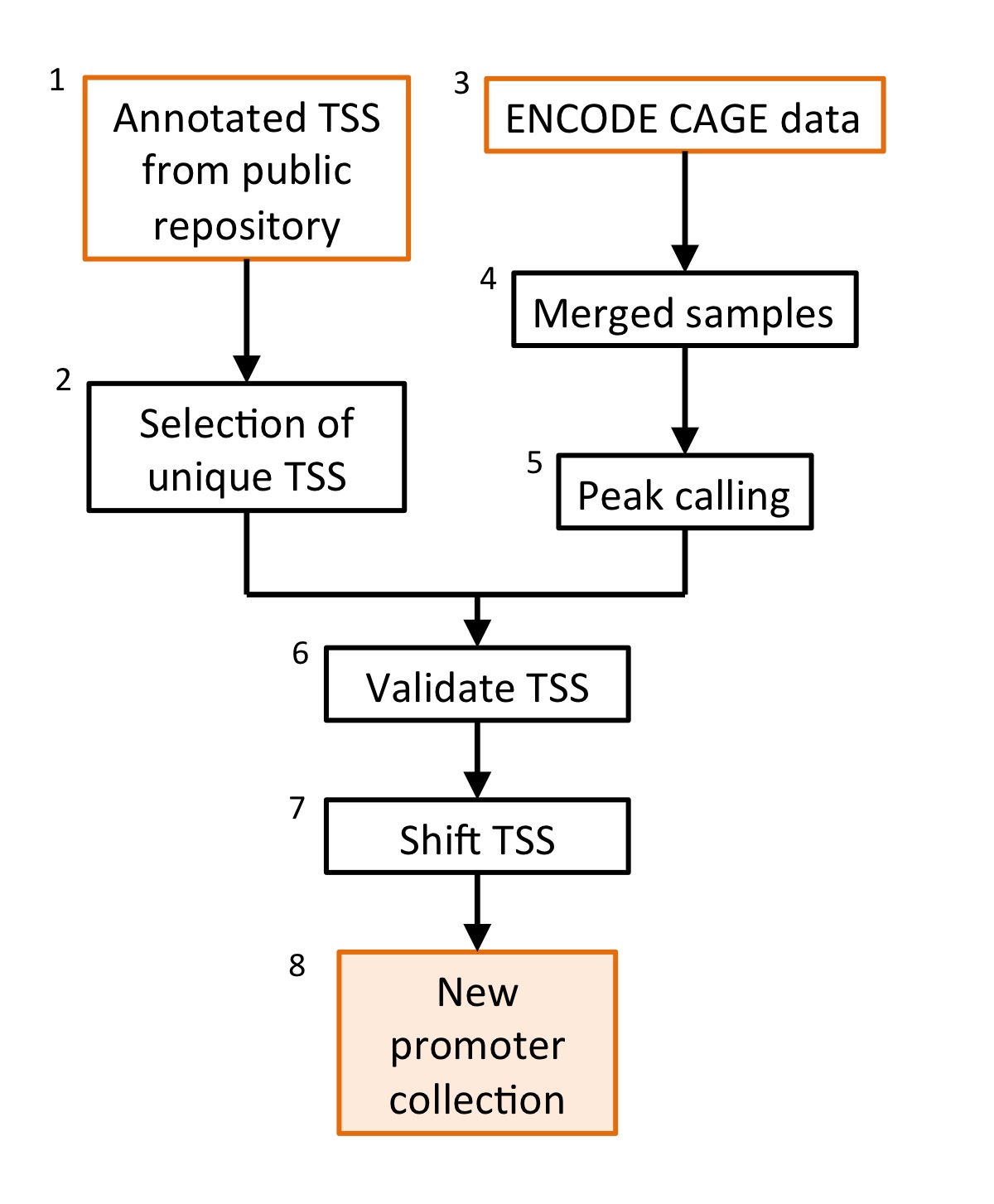
|
Figure 1: flowchart describing how to make a validated
promoter collection. Each step will require the following:
|
1. Make a validated promoter collection
1.1 Make a promoter collection from UCSC Genome Browser data
The first step in this tutorial is to download a promoter collection from a publicly available database. Here it is shown how to do it for UCSC gene annotation table but you can do it in BioMart as well. This is your starting point: a promoter collection that is not experimantally validated, with a low accurancy in placing the TSS. You will then use CAGE data to validate the promoters (selecting the promoters that are expressed) and to shift the TSS location to the nearest CAGE peak. This should represent a better estimate of the true TSS location.
-
To download the promoter collection go to UCSC table
browser and download UCSC gene annotations:
-
Select:
clade: Mammal
genome: Human
assembly: hg19
group: Genes and Gene Predictions
track: UCSC Genes
table: knownGene
output format: selected fields from primary and related tables
Click "Get output" - Select all the fields of the table hg19.knownGene plus geneSymbol from hg19.kgXref table and save the output to a file (for example ucscHumanGenes.txt)
-
Select:
-
This tab-delimeted file can't be used as input for the ChIP-Seq
tools. You need to convert it into an SGA
file. Here is an R code that can be used to generate a promoter
SGA file from the UCSC table (lines starting with
a
#symbol are comments):# Read file with transcript coordinates into a table
ucscGenes <- read.table("ucscHumanGenes.txt", as.is=T, sep="\t", header=F)
# Have a look at the first few lines of the table:
head(ucscGenes)
# Get the TSS position for each transcript: it is the 4th field
# (transcript start position) if the transcript is mapped to the +
# strand; or the 5th field if the transcript is mapped to the -
# strand. [Note that you have to add 1 to the 4th field since UCSC
# starts chromosomes at base 0 whereas SGA starts at 1]
tss <- ucscGenes[,4] + 1
tss[which(ucscGenes[,3] == "-")] <- ucscGenes[which(ucscGenes[,3] == "-"),5]
# Make a table with similar layout as SGA file:
# Chromosome Feature Position Strand Count ID
sga <- cbind(ucscGenes[,2], "TSS", tss, ucscGenes[,3], 1, ucscGenes[,13])
head(sga)
# There are multiple transcripts that share the same TSS, get only
# one copy:
sga.unique <- unique(sga)
# Order the SGA file by chromosome, position and strand:
sga.order <- sga.unique[order(sga.unique[,1], as.numeric(sga.unique[,3]), sga.unique[,4]),]
# Write out the final SGA to a file:
write.table(sga.order, "ucscHumanGenes.sga", col.names=F, row.names=F, sep="\t", quote=F, eol="\n")
1.2 Validate the UCSC promoters with CAGE data
To start the validation process (point 1 in the next list), you have to extract CAGE peaks from the genome. To do so, you will use samples from the ENCODE CAGE data . To validate the largest number of promoters, you should use a sample that is the result of merging all longPolyA samples (mRNA enriched samples) that are present in this serie. The merged sample is available from our server in the drop-down menu. The resulting peaks will then be used to validate and select promoters (point 2) for subsequent analysis.
-
Use ChIP-Peak to find peaks in ENCODE CAGE data
using the following parameters. You can try and change them. For
example changing the 'Peak Threshold' would affect the
sensitivity of your analysis, lower values produce more peaks
that can be used to validate more TSSs. Reducing the 'Window
Width' and 'Vicinity Range' would affect broad peaks,
potentially splitting them in smaller peaks.
- Select the following sample:
- Genome: hg19
- Data Type: ENCODE RNA-seq
- Series: GSE34448, CAGE data from ...
- Sample: All samples cell longPolyA
- Set the following parameters:
- Strand : oriented
- Window Width (bp) : 200
- Vicinity Range (bp) : 200
- Peak Threshold (nb of reads) : 50
- Count Cut-off (nb of reads) : 99999999
- Peak Refinement : on
- Run the job.
- Save the SGA file with the peaks.
-
Use ChIP-Cor to select promoters with CAGE peaks
nearby:
- Use the UCSC human TSS file (generated with R before) as reference feature (using the 'Upload custom Data'). It is an SGA file, mapped on hg19 and is an oriented feature.
- Use the ENCODE CAGE peaks file you have just saved as a target feature (again with the 'Upload custom Data'). Select the "+" strand only. This way, you will get the distribution of CAGE peaks that are near UCSC annotated TSS that map to the sense strand relative to the TSS orientation.
- Reduce the range by a factor of 10: from -100 to 100; Window Width to 1 and Count Cut-off to 1.
- You are now ready to submit the analysis.
-
In the result page use the "Enriched Feature Extraction
Option" to extract promoters that have at least one CAGE
peak near them (in a window of 200 bp around them). To do
so, set the following parameters:
From -100; To 100; Threshold: 1; Cut-Off : 1; set "Ref Feature Oriented " as ON - You can save the SGA file with the validated promoters
1.3 Shift the position of the annotated TSS to a CAGE peak nearby
At this point in the analysis you have a promoter collection that has been validated by CAGE data with the original TSS coordinates from UCSC. These are not very precise. To increase the TSS precision you could shift them to the nearest CAGE peak position or, alternatively, shift their location to the nearest base with the higher CAGE count. These two values do not always coincide, since ChIP-Peak return the 'center of gravity' of the peak area, and, expetially for broad peaks, it might be away from the base with the higher CAGE count. [Remember that CAGE reads represent the 5-end of mRNA molecules and so the transcription start site of a gene.] We found that the second option gives better results in term of quality. In this section you will learn how to do it. This is a two step procedure in which you [1] extract the position of the CAGE tags around each promoter and [2] shift the TSS to the nearest CAGE maximum.
- Use ChIP-Extract to extract CAGE tags around these promoters
- You can used the link to ChIP-Extract from the previous page or alternatively upload the file with validated promoters as reference feature. As usual, promoters are oriented features.
- Use the ENCODE samples you used at the beginning of this tutorial (All samples cell longPolyA). Select "+" strand only.
- Set the range from -100 to 100; Window Width 1 and Count Cut-off 9999999999
- Submit the job
- Save the "Table (TEXT)" file. This matrix stores the distribution of CAGE tags around each promoter.
-
Use R to shift promoters to the nearest CAGE tag maximum using
the following code. Here
ucsc_encodeCageTags.outis the matrix file you just generated anducscHumanGenesActive.sgais the list of validated promoters.# Read the matrix file:
cage <- read.table("ucsc_encodeCageTags.out")
# The function getShift calculates the distance (= shift) between the TSS and
# the CAGE maximum. It accepts a vector (x) and return the shifting distance
# from the midpoint.
getShift <- function(x){
maxCage <- which(x == max(x))
min(maxCage - ceiling(length(x)/2))
}
# Apply the function to each row of the cage matrix: get the shift position
# for each promoter in your collection
promoterShift <- apply(cage, 1, getShift)
# Read the SGA file with the list of active genes (it has the same order as
# cage matrix)
ucscGenesActive <- read.table("ucscHumanGenesActive.sga", as.is=T, sep="\t", header=F)
# Shift promoters to the nearest cage maximum using the shift values you have
# calculated:
ucscPromotersShift <- ucscGenesActive
ucscPromotersShift[,3] <- as.numeric(ucscPromotersShift[,3]) + promoterShift
# Write out the result as a SGA-formatted file:
write.table(ucscPromotersShift, "ucscPromotersShift.sga", col.names=F, row.names=F, sep="\t", quote=F, eol="\n")
Now you have an experimentally validated promoter collection with the TSS position defined as the position of CAGE maximum. The next tutorial will show you how to check the quality of your collection and how to compare it to the initial UCSC annotation and EPDnew. Please note that after shifting there is a high probability that your collection is not properly sorted. If you want you can sort it with following bash command:
sort -s -k1,1 -k3,3n -k4,4 ucscPromotersShift.sga >
ucscPromotersShiftSorted.sga
Or, alternatively, switch-on the sorting option when uploading your collection on the server.
2. Quality control of your promoter collection
In this second section of the tutorial you will perform some quality controls on your promoter collection to see if it is any better than UCSC original collection and to compare it to EPDnew. First you will check the motif distribution around promoters and then some histone marks.
Motif search around promoters is done using OProf tools (part of the SSA web server). It accept input in several file formats such as SGA, even if the internal file type is called FPS. Start the analysis as follow:
- Use OProf to generate a density distribution plot of core promoter element around promoter lists
- Upload your SGA file (select hg19 as genome)
- Select TATA-box from the motif library "Promoter Motifs"
- Set the range from -99 to 100, Window size 16, Window shift 1 and Search mode forward
- Run the job and save the Text (numerical) file
- Do the same with the Initiator motif changing the Window size to 8
- Repeat the same analysis for the UCSC full promoter list that you have generated at the beginning of this tutorial and for EPDnew 003 (EPDnew can be found in the available dataset under "Genome annotation" data type) each time saving the TXT file with the intensities.
-
Now use R to compare the databases:
-
Use R to plot and compare the distribution of TATA-box and
Initiator motifs around each promoter list (your, UCSC and
EPDnew). The R code for the TATA-box is presented here:
# As usal start by reading the data into R objects for each promoter
# collection:
epdnewTata <- read.table("EPDnew_tata.dat")
myTata <- read.table("my_tata.dat")
ucscTata <- read.table("ucsc_tata.dat")
# Then simply plot them together to a JPEG file:
jpeg("tata-box.jpg")
plot(epdnewTata[,1], epdnewTata[,2], type="l", main="TATA-box", xlab="Distance from TSS", ylab="Frequency")
points(myTata[,1], myTata[,2], type="l", col="red")
points(ucscTata[,1], ucscTata[,2], type="l", col="green")
legend("topright", legend=c("EPDnew","My collection","UCSC"), col=c("black","red", "green"), lty=1)
dev.off()
- Do the same for the Initiator
-
Use R to plot and compare the distribution of TATA-box and
Initiator motifs around each promoter list (your, UCSC and
EPDnew). The R code for the TATA-box is presented here:
-
Use ChIP-Cor to see the distribution of the
histone mark H3K4me3 (an active mark present in promoters) in
the 3 databases:
- Use one of the tree databases as reference feature (EPDnew can be found in the available dataset under "Genome annotation" data type). Remember, promoters are oriented features.
- As target feature you can use H3K4me3 sample from Barski 2007 under "ChIP-seq" data type. Set strand "any" and centering "70"
- Run the job and save the Text numerical file as before
- Use R to draw a common picture (as before)
Considering motif plots and histone marks analysis, how does your database compare to the other two? Write a short document reporting your findings (parameters used, number of promoters you have found, ...) and the figures you just generated.
3. Promoter selection
In this final section you will learn how to select promoters that are expressed in a particular sample. You will check their chromatin status and compare it to non-expressed promoters.
As an example, you will use the ENCODE data for cell line GM12878. This is an ENCODE tier 1 cell line. As a consequence, it has been heavily studied, providing data for almost all conditions / targets used by the consotium.
In a first step you will use ChIP-Cor to select promoters that are expressed in GM12878, then you will study their histone marks distribution and additionally check their Gene Onthology (using the GREAT suite)
-
Use ChIP-Cor to select promoter expressed in GM12878
- Upload your promoter collection as a reference feature. As usual, this is an SGA file mapped on hg19 and promoters are oriented features. Switch-on the sorting option.
-
Select the following target feature:
- Genome : hg19
- Data Type : ENCODE RNA-seq
- Series : GSE34448, CAGE data from ...
- Sample : GM12878 cell longPolyA rep1
- Strand : "+"
- Set the range from -100 to 100; Window Width 1 and Count Cut-off 9999999999
- Submit the job
-
Use the "Enriched Feature Extraction Option" tool to extract
promoters with high (expressed) and low (non-expressed) tag
count around them:
- From -100 to 100
- Threshold : 100
- Cut-Off : 9999999999
- Switch to Depleted Feature Extraction : Off (this will extract expressed promoters)
- Ref Feature Oriented : On
- Run the analysis and save the SGA file
- You can use the link to the GREAT suite in this page to get a Gene Onthology enrichemnt of the expressed genes.
- Repeat the same analysis turning On the "Switch to Depleted Feature Extraction" to extract non-expressed promoters.
-
Use ChIP-Cor for studing the histone marks distribution around
these two set of promoters:
- As reference feature choose one of the two sga file you just saved (then repeat the analysis with the other).
- Check all GM12878 specific histone marks / variants in the serie "GSE29611, Histone modification ..." under "ENCODE ChIP-seq" Data Type. You can use "any" strand with a centering of "100".
- Which histone mark / variant is predominant around expressed promoters? Which around non-expressed? Does this make sense?