EPD's 40th Anniversary Symposium is open for registration !!! (click here)
(taking place in Geneva on Sep 4 2026, just after
ECCB 2026,
for more information, see https://epd.expasy.org/EPD_symposium2026/)
TSS assembly pipeline for Hs_EPDnew_006
Introduction
This document provides a technical description of the transcription start site assembly pipeline that was used to generate EPDnew version 006 for H. sapiens.Source Data
Promoter collection:
| Name | Genome Assembly | Promoters | Genes | PMID | Access data | ||
|---|---|---|---|---|---|---|---|
| Gencode | Dec 2013 GRCh38/hg38 | 35320 | 17056 | 27250503 | SOURCE | DOC | DATA |
Experimental data:
| Name | Type | Samples | Tags | PMID | Access data | ||
|---|---|---|---|---|---|---|---|
| FANTOM5 | CAGE | 941 | 18,244,201,540 | 24670764 | SOURCE | DOC | DATA |
| ENCODE | CAGE | 145 | 7,134,200,060 | 22955620 | SOURCE | DOC | DATA |
| ENCODE | RAMPAGE | 225 | 13,540,041,874 | 22936248 | SOURCE | DOC | DATA |
Assembly pipeline overview
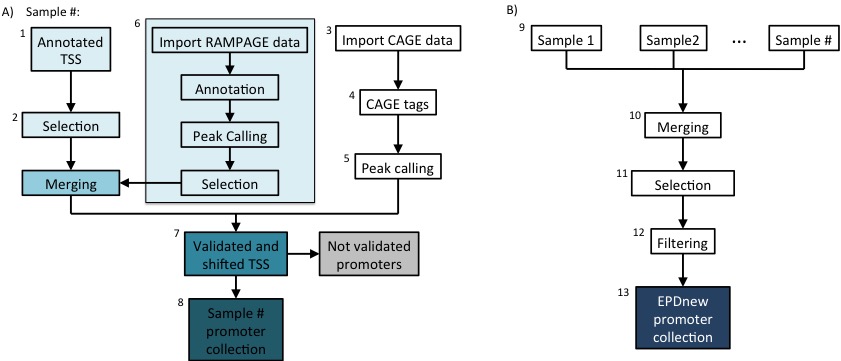
|
Description of procedures and intermediate data files
1. Download of annotated promoters
Data for the latest reference Human GENCODE release (v28) was
downloaded from EBI FTP website. Transcrips were kept if they
belong to a protein coding genes (flag 'gene_type' =
'protein_coding') and the transcript support level equal to 1 (all
splice junctions of the transcript are supported by at least one
non-suspect mRNA, this is the most stringent level).
Gene names were taken from the field "gene_name". Since
the EPD format doesn't allow gene names longer than 18 characters,
we checked whether the names repsected this limitation.
Transcripts with the same TSS position were merged under a common
transcript ID. As a consequence, the total number of TSS in the
list was 35320 covering 17056 protein coding genes.
2. Gencode TSS collection
The Gencode TSS collection is stored as a tab-deliminated text file conforming to the SGA format. The six fields in the file contain the following kinds of information:
- NCBI/RefSeq chromosome id
- "ENSEMBL"
- position
- strand ("+" or "-")
- "1"
- TranscriptID..GeneName.
3 Import Single-end sequencing data: CAGE
CAGE Tag Data were downloaded from UCSC ftp-site and FANTOM5
http-site (see links above). The source files are in bam format
mapped on hg19 genome assembly. Samples were lifted-over to hg38
genome assembly using the liftOver tool. The complete list of
files can be found
here for ENCODE and
here for FANTOM5. Bam files were converted into bed files
with bamToBed program. Files were kept and analysed
individually.
4. CAGE mapping data
The compressed versions of these files are available from the MGA archive (see links above).
5. CAGE peak calling
Peak calling for each individual CAGE and RAMPAGE data file has been carried out using ChIP-Peak on-line tool with the following parameters:- Window width = 1
- Vicinity range = 200
- Peak refine = N
- Count cutoff = 9999999
- Threshold = 5
6. RAMPAGE data analysis
RAMPAGE is a TSS mapping technique that uses paired-end sequencing to uniquely assign a 5'-end tag (first read in the pair) to an annotated gene using the 3'-end tag (the second read in the pair). This gives high confidence when assigning TSS to genes when they map outside gene boudaries (potentially very far from them). The new nature of the data required a specific analysis and a modification in the general EPDnew validation pipeline. As for CAGE data, each RAMPAGE sample was analysed separately and merged together only at the end to generate a RAMPAGE-specific promoter collection. The new analysis is chararacterised for the following steps: (1) Data import and annotation; (2) Peak calling (3) Peak selection and (4) Quality control. Each step will be brefely described.
Data import and annotation
Data was downloaded from the ENCODE web-site as BAM files mapped
on GRCh38/hg38 genome assembly and converted into SGA format
using the following procedure:
- Keep reads that have a good mapping score (5th field in the bed file = 255)
- Split the reads in a pair in to two files and keep the read ID
- Annotate the second read file if they map inside a gene exon and have the same orientation
- Annotate the first read file using the read ID
- Discard reads if not annotated
Peak calling
Peak calling was carried out as described for CAGE data. Since
RAMPAGE tags were already annotated to a specific gene, RAMPAGE
peaks retained the gene annotation given to the tags that
composed them.
Peak selection
Annotated RAMPAGE peaks were selected if they map outside the
gene boudaries they belong to.
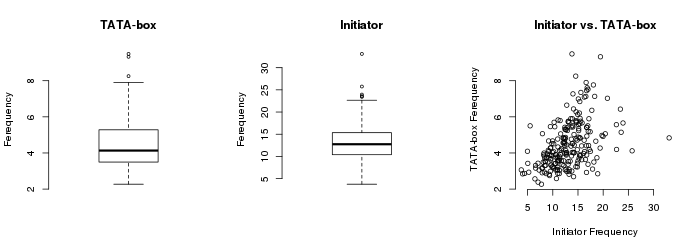
Quality Control
Each sample-specific peak collection (225 TSS collections) was
quality checked using the usual EPD QC parameters: density
distribution of known Core Preomoter Elements (CPEs) around
putative TSS. For this analysis we used two well known CPEs, the
TATA-box and Initiator (Inr) that are
found at position -29 and 0 relative to the TSS and mesured
their frequencies in each of the 225 RAMPAGE-derived TSS
collections. Sample specific TATA-box and Initiator frequencies
are summarised in the following figure:
|
7. Gencode+RAMPAGE TSS validation dy CAGE data
Each sample in the collection (CAGE peaks and Gencode+RAMPAGE TSS) was then processed in a pipeline aiming at validating transcription start sites with CAGE peaks. A Gencode+RAMPAGE TSS was experimentally confirmed if a CAGE peak lied in a window of 200 bp around it or if mapped in the 5'UTR region of an annotated gene and if it had a maximum high of at least 5 tags (50 tags for peaks in the 5'UTR). The validated TSS was then shifted to the nearest base with the higher tag density. Secondary promoters for genes with multiple TSSs were discarded if their expression level was below 10% of the strongher gene-specific initiation site or below 10 tags.
8. Promoter collection for each sample
Each sample in the dataset was used to generate a separate promoter collection. Potentially, the same transcript could be validated by multiple samples and it could have different start sites in different samples. To avoid redundancy, the individual collections were used as input for an additional step in the analysis (Assembly pipeline part B).
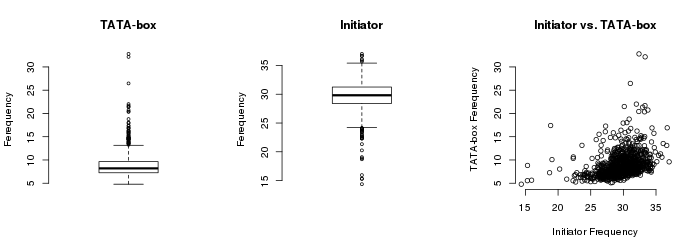
9. Quality controls of sample-specific promoter collections
The quality of promoter collections derived from each sample was tested to exclude low quality samples from the final collection. To achive this, each promoter collection was scored according to the distribution of the TATA-box and Inr motif in the expected position (-29bp from the TSS and at the TSS respectively). Samples with very low motif frequencies (Inr frequency < 10% and TATA-box < 5%) were discarded (3 samples in total) from further analyses. The figure below shows the distribution of TATA-box and Inr in all sample-specific promoter collections:
|
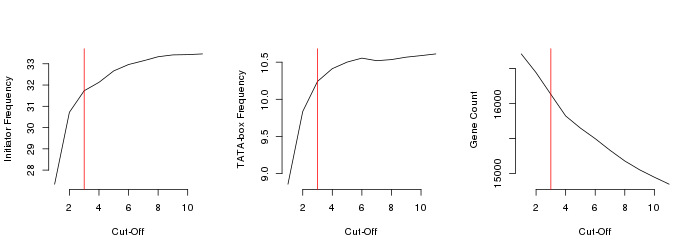
10. Merging collections and further TSS selection
The good-quality promoter collections were merged into a unique file and further analysed. The promoter of a transcript was mantained in the list only if validated by at least 3 samples. We chosed this Cut-off value since it ensured a good increase in Inr and TATA-box friquency without affecting too much the total number of validated genes:
|