ChIP-Seq Analysis Tutorial
Overview
In this tutorial, we will try to give step-by-step examples/exercises on how to use our ChIP-seq methods to analyse transcription factor binding sites, histone modifications, and CAGE data. The tutorial will be organized in three parts dealing with:
- A. ChIP-Seq Data Analysis : locating in vivo STAT1-binding sites
- B. ChIP-Seq Data Analysis : analyzing histone modifications
- C. Higher-Level Analysis of ChIP-Seq Data
IntroductionChIP-Sequencing, also known as ChIP-Seq, is mainly used to analyze protein-DNA interactions. ChIP-Seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the set of cis-acting targets of DNA-associated proteins or factors on a genome scale. It can be used to precisely map global binding sites for any protein of interest. Previously, ChIP-on-Chip was the most common technique used to identify trascription factor DNA interactions. Chip-Seq is also used to study epigenetic events such as histone modifications and DNA methylation. This epigenetic information is complementary to genotype and expression analysis. ChIP-Seq technology is rapidly replacing ChIP-on-Chip which requires a hybridization array. ChIP-on-Chip necessarily introduces some bias, as an array is restricted to a fixed number of probes, whereas sequencing is thought to have less bias, although the sequencing bias of different sequencing technologies is not yet fully understood. Specific DNA sites in direct physical contact with transcription factors and other proteins can be isolated by chromatin immunoprecipitation. ChIP produces a library of DNA sites bound to a probe in vivo. Massively parallel sequence analyses are used in conjunction with whole-genome sequence databases to analyze the interaction pattern of any protein with DNA, or the pattern of any epigenetic chromatin modifications. This can be applied to the set of ChIP-able proteins and modifications, such as transcription factors, polymerases and transcriptional machinery, structural proteins, protein and histone modifications. Since the data are 'high-quality' sequence reads, ChIP-Seq offers a rapid analysis pipeline (as long as a high-quality genome sequence is available for read mapping) for Protein-DNA association studies as well as epigenetic genome organization.
|
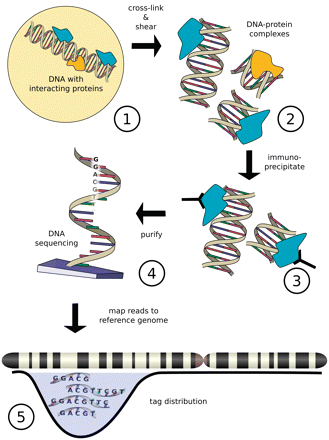 Figure from Szalkowski, A.M, and Schmid, C.D.(2010).
Figure from Szalkowski, A.M, and Schmid, C.D.(2010). Rapid innovation in ChIP-seq peak-calling algorithms is outdistancing banchmarking efforts. Briefings in Bioinfomatics. |
In ChIP-seq analysis, the input is a list of sequence read (or tag) start coordinates mapped to the genome. As an internal working format we propose a simplified GFF format that we have called SGA (Simplified Genome Annotation). The ChIP-Seq tools use SGA files as INPUT and OUTPUT. SGA files are used to represent ChIP-Seq data as well as other genome annotations such as the location of TSSs or matches to consensus sequences.
SGA is a single-line-oriented and tab-delimited format with the following six fields:
- Sequence name (Char String)
- Feature (Char String)
- Sequence Position (Integer)
- Strand (+/- or 0)
- Tag Counts (Integer)
- Comment/Description (Char String)
The latter is an optional field.
Users can either utilise a set of server-resident SGA files from selected public experimental data (ChIP-Seq Experiments) or upload their own SGA file. GFF and FPS are also allowed formats. In particular, we will use FPS files for motif analysis with SSA programs, which are a set of analysis tools that we have developed to study sequence motifs that occur at characteristic distances upstream or downstream from a functional site in a nucleic acid sequence. For an introduction to SSA, please refere to the on-line SSA Tutorial. In particular, we will use the OProf, SList and PatOp programs.
For a more detailed description of the ChIP-Seq tools and data formats, please refer to the ChIP-Seq Technical Document.
