ChIP-Seq Analysis TutorialPart 2: ChIP-Seq Data Analysis
|
|
In this section, we will extend the analysis of ChIP-Seq data to:
B. Analyzing histone modifications
Both the Barski and Mikkelsen data are available on our ChIP-Seq Web site and we are now going to present a few examples of histone autocorrelation studies (using the ChIP-Cor tool) along with genome partitioning (by means of the ChIP-Part tool).
Methodology
A typical genome partitioning analysis involves the following steps:
- Perform correlation studies with ChIP-Cor to evaluate the average fragment size of the ChIP-Seq data under study, and to compute the average count density in signal-rich and signal-poor regions.
- Partition the genome into signal-rich and signal-poor regions with ChIP-Part.
- The tag count density threshold used as a parameter for the genome partitioning algorithm is set to be twice the average tag count density outside the enriched regions.
1. Correlation Studies (ChIP-Cor)
We have seen in the previous example that ChIP-Cor can be used for studying
the distribution of 5'-3' tag reads positions and that this gives us an estimate of the average DNA fragment size that has been precipitated by ChIP.
In addition to that, ChIP-Cor is also useful to generate auto-correlation plots for different histone modifications.
Let's take as an example two different histone methylation marks in embryonic mouse cells (ES): H3K6 trimethylation and H3K4 trimethylation.
Go on our ChIP-Seq Web server at ChIP-Cor, and follow the instructions below:
|
You are now ready to submit the job.
Do the same for H3K4me3 .
If you compare the two auto-correlation plots for H3K36 and H3K4, you see that H3K36 shows long range correlation whereas H3K4 has short range correlation. This indicates that certain epigenetic modifications are less localized than others and bind DNA over larger regions, producing relatively broad peaks. An alternative hypothesis would be that the predominant location of H3K4me3 is close to chromosome ends (telomers). We can observe that count densities in enriched regions are 0.012 and 0.07 for H3K36 and H3K4 respectively which correspond to approximately 2 and 14 counts per nucleosome, assuming that the whole length of DNA per nuclesome is about 150-200 bps. Average count densities in signal-depleted regions are 0.005 and 0.007 for H3K36 and H3K4 respectively.
Note that with ChIP-cor you can download the numerical results as a text file which is provided as a link underneath the histogram plot. This is useful if one wants to overlay more than one plot.
As we have already mentioned, Chip-Cor is also used to study the fragment size distribution of ChIP-seq data. To this purpose, we want to compare 5'-3' correlation of H3K4me3 regions from Mikkelsen et al. with H3K4me3 from Barski et al.. As said in the previous introduction, contrary to the Mikkelsen data, the Barski data have single-nucleosome resolution.
Go to ChIP-Cor and follow the instructions below:
|
You are now ready to submit the job.
You can perform the same anylisis for the Mikkelsen et al. data.
As you can see from the distributions obtained from ChIP-Cor (Barski H3K4me3 and Mikkelsen H3K4me3), Mnase treatment in combination with the ChIP will lead to nucleosome-sized DNA (about 147 bp) that can be sequenced by high throughput sequencers, resulting in a fine mapping of the H3K4me3 pattern in the genome.
2. Using partitioning for analyzing differential histone modification analysis (ChIP-Part)
The partitioning model is more appropriate than peak recognition for the detection of regions or peaks of higly variable size and for differential chromatin state analysis. Histone modifications are the result of mechanisms that spread across the genome.
For example, H3K36me3 is the result of trascriptional activity that leads to DNA methylation, whereas the modification H3K4me3 is associated with the transcription start site of active genes.
Our partitioning algorithm (see also the ChIP-Seq Technical Document) uses a fast dynamic programming approach to maximize a global scoring function in order to split the genome in signal-rich and signal-poor regions.
We remind that the scoring function is defined by means of two parameters, the count density threshold and the transition penalty, as the sum of scores of:
- Transitions (penalty)
- Signal-rich region scores: length x (local count-density - threshold)
- Signal-poor region scores: length x (threshold - local count-density)
We will use the ChIP-Part tool to compare H3K4me3 chromatin domains in four different mouse cell lines from Mikkelesen et al.: embryonic stem cell lines(ES and ESHyb), embryonic fibroblasts (MEF), and neural progenitors (NP).
Go to the program ChIP-Part (ChIP-Part stays for ChIP-Seq Partitioning tool) and procede as follows:
|
As a rule of thumb., you can set the count density threshold to twice the average tag count density outside peak regions.
As you can see, ChIP-Part for supported species generates the following output:
- A default two-line-oriented SGA file representing signal-reach DNA regions;
- GFF, FPS, BED formats;
- A link is generated to automatically display the results on the UCSC browser as a custom annotation track;
- A statistical report with the following information:
- Total number of processed sequences, total DNA length, and total number of fragments;
- Total length of fragments, average fragment length, and percentage of total DNA length;
- Percentage of total counts, average number of counts, and number of count per bp (count density).
In our case, we get 24402 fragments of average length 1778 bp.
If you click on the hyperlink to UCSC you can view the results of the partitioning program in the UCSC genome browser. On our ChIP-Seq Web server we make available a list of URLs for viewing public ChIP-Seq data as custom tracks within the UCSC genome browser. These custom tracks are provided in bigWIG format and their URLs are specified at
So, you can upload the track for H3K4me3 in mouse ES cells from Mikkelsen et al. and compare it with the results from the partitiong program. Here is an example of such a comparison for H3K4me3 in mouse ES cells:
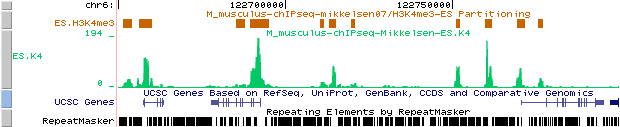
Custom tracks:
Mikkelsen et al. : results of ChIP-part program (ES.K4 BED file)
ES.K4 from : http://ccg.vital-it.ch/bigWIG/mikkelsen07/ES.K4_m.BW
As we can see from these results, our partitioning model seems to fit well the data from Mikkelsen et al..
It is often useful to save the BED file generated by the partitiong program for further analysis (ES.K4.bed).
Repeat the same analysis for H3K4 in ESHyb, MEF, and NP cell lines by adjusting the count density threshold accordingly, and save the results in BED format.
If we display the H3K4me3 profiles from Mikkelsen et al. on the UCSC browser, we can see how chromatin domains are differentially occupied in different cell lines.
Go to UCSC and proceed as follows:
- Select Assembly : "Mouse" "July 2007 (NCBI37/mm9)";
- Select Position : chr6:122,650,000-122,800,000;
You then upload the Mikkelsen maps of chromatin state that we make available on our ChIP-Seq server (in bigWIG format) by doing the following:
- Click the "add custom tracks" button;
- Paste the URL(s)s of the data file(s) from Mikkelsen et al. (ex: http://ccg.vital-it.ch/bigWIG/mikkelsen07/ES.K4_m.BW) in the box provided and click submit;
Here is how the Mikkelsen annotation tracks look like on the UCSC genome browser:
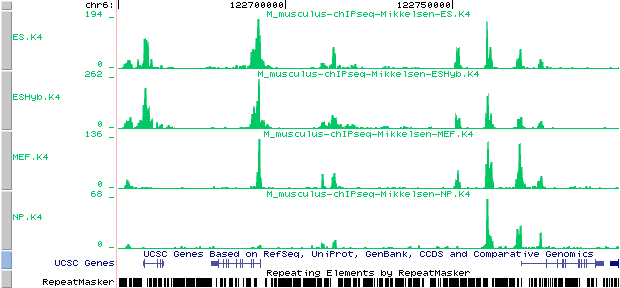
Custom tracks:
Mikkelsen et al. (2007). Genome-wide maps of chromatin state in pluripotent
and lineage-committed cells. Nature 448, 553-560
ES.K4 from : http://ccg.vital-it.ch/bigWIG/mikkelsen07/ES.K4_m.BW
ESHyb.K4 from : http://ccg.vital-it.ch/bigWIG/mikkelsen07/ESHyb.K4_m.BW
MEF.K4 from : http://ccg.vital-it.ch/bigWIG/mikkelsen07/MEF.K4_m.BW
NP.K4 from : http://ccg.vital-it.ch/bigWIG/mikkelsen07/NP.K4_m.BW
Finally, we can upload the tracks from the paritioning analysis as we have previously shown.
Here are the results displayed on the UCSC browser:
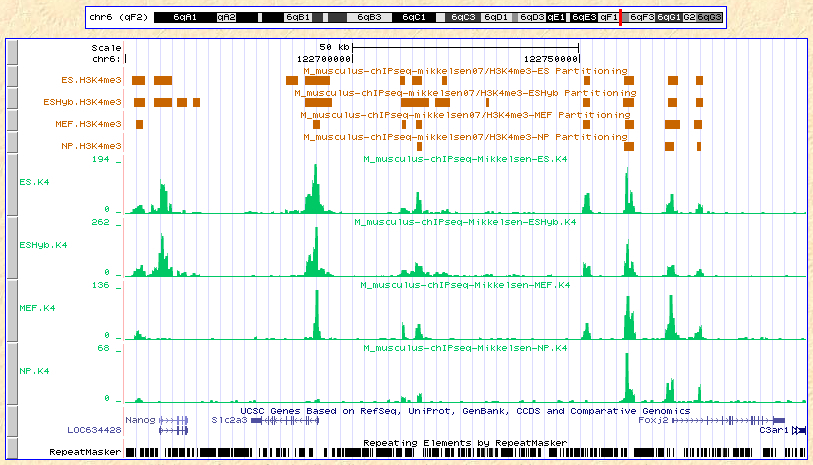
Custom tracks:
Partitioning data:
ES.K4.bed,
ESHyb.K4,
MEF.K4,
NP.K4.
Mikkelsen et al. (2007). Genome-wide maps of chromatin state in pluripotent
and lineage-committed cells. Nature 448, 553-560
As we can see from the results above, the ChIP-part algorithm is consistent with an intuitive interpretationof the 'count/tag' profiles of our data.
