
EPD's 40th Anniversary Symposium is open for registration !!! (click here)
(taking place in Geneva on Sep 4 2026, just after
ECCB 2026,
for more information, see https://epd.expasy.org/EPD_symposium2026/)
- PWMTools
- Browse and Download PWMs
- MGA Database
- Other Resources
- References
- Frequently Asked Questions
- What is new
PWMEval-Selex - Evaluation of Binding Models based on SELEX/SELEX-like data
  |
Background |
SELEX uses purified proteins to select high-affinity binding sites from random libraries in vitro. By utilizing new sequencing technologies it is possible to derive binding energy profiles from SELEX methods quite efficiently using a method called high-throughput SELEX (HT-SELEX). HT-SELEX consists of several cycles of incubating the DNA-binding protein with a mixture of DNA sequences, enrichment of the bound DNA sequences, sequencing a sample of them and feeding them to the next cycle. An advantage of this approach is that the output (the number of counts observed for each sequence) is digital and there is a sufficient depth of data to allow use of sophisticated staistical methods that provide more accurate models of protein DNA-binding specificity than previously available.
SMiLE-seq is a new technique for the characterization of DNA-binding proteins in a much faster, more accurate and efficient way. The core of SMiLE-seq is a microfluidic platform that involves capillary loading of in vitro-transcribed and -translated bait TFs, and target double-stranded DNA from a pool of random sequences. The transcription factor (TF) is bound to the surface of the microluidic device by antibodies and some fraction of the DNA binds to the TF. The unbound DNA is expelled by washing, so the bound fraction can be measured. Bound DNA is subjected to high-throughput sequencing and a hidden Markov model (HMM)-based TF motif discovery pipeline for de novo identification of DNA-binding specificities and affinities of different families of full-length TFs and TF dimers.
For each sequence, the sum occupancy score is calculated, which is the sum over all motif matches weighted by their respective scores. The sum occupancy score is used to rank all sequences.
Shuffled sequences are used as the negative set.
By default, the top-ranked 50% of the sequences are used for testing the PWM model.
The PWM format used is the letter-probability matrix.
For probe sequence s and matrix PWM of length k, the sum occupancy score is computed as follows:
|
|
(1) |
where PWMi(s) is the probability of base s in position i of the PWM, and pi is the nucleotide prior (or background) probability. PWM frequencies are normalized by the background sequence composition.
The background composition normalization can be computed by the following three methods:
- based on a uniform background sequence composition (uniform i.e: 0.25);
- based on the nucleotide composition of the entire sequence library (library comp);
- based on the nucleotide composition of each single sequence (sequence-based).
Scores of binding predictions are reported in AUC of the receiver operating characteristic curve (ROC).
The motif libraries have been downloaded from the MEME Motif Database, and include the following sets: JASPAR CORE 2018, Human and Mouse HT-SELEX motifs (Jolma 2013), UniPROBE Mouse, and HOCOMOCO (version 11).
1.1 Conversion of position frequency or count matrix (PFM, JASPAR, TRASFAC) to letter probability matrix (LPM)
The conversion of base counts to corrected frequencies (fib), that is relative frequencies which are corrected by pseudo-count fractions distributed according to residue priors, uses the following formula:
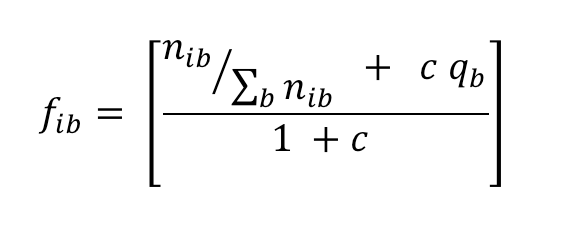
|
(2) |
1.2 Conversion of position weight matrix (PWM) to letter probability matrix (LPM)
A position weight matrix can be converted to a letter probability matrix in two ways:
- by using a cut-off value and a given background base composition, we generate all nucleotide sequences represented by the weight matrix and the cut-off and compute the base frequencies,
- or by inverting the log likelihoods with the following formula:
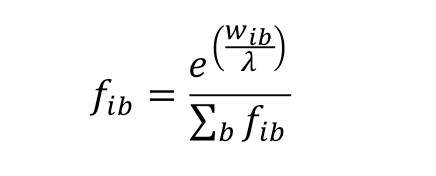
|
(3) |
|
|
Matrix Formats |
TRANSFAC-style matrices look like this:
AC M00223 XX ID V$STAT_01 XX DT 29.11.1995 (created); ewi. DT 11.03.2003 (updated); dtc. CO Copyright (C), Biobase GmbH. XX NA STATx XX DE signal transducers and activators of transcription XX BF T01575 STAT1alpha; Species: mouse, Mus musculus. BF T01492 STAT1alpha; Species: human, Homo sapiens. BF T01573 STAT1beta; Species: human, Homo sapiens. XX PO A C G T 01 0 0 0 14 T 02 0 0 0 14 T 03 2 12 0 0 C 04 0 12 0 2 C 05 0 9 2 3 C 06 4 2 8 0 G 07 0 0 8 6 K 08 13 1 0 0 A 09 14 0 0 0 A XX BA 14 genomic sequences of 14 different genes XX CC compiled sequences XX RN [1]; RE0003481. RX PUBMED: 7774815. RA Horvath C. M., Wen Z., Darnell jr J. E. RT A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain RL Genes Dev. 9:984-994 (1995). XX
JASPAR-style matrices look like this:
>MA0137.2 STAT1 A [ 208 859 251 10 8 106 23 528 696 53 1900 2030 954 336 417 ] C [1076 496 574 22 14 1921 1900 762 31 30 124 17 263 760 804 ] G [ 415 279 144 11 38 14 7 115 1292 1700 29 23 552 270 425 ] T [ 378 446 1112 2038 2023 44 155 680 66 302 32 15 315 714 431 ]
PFM-style matrices look like this:
> M00224 STAT1 8.0 18.0 41.9 32.1 23.2 9.7 37.2 29.8 13.9 31.3 19.8 35.2 17.5 37.9 38.1 6.4 59.6 13.5 15.1 11.9 31.2 26.3 9.4 33.2 0 0 0 100 0 0 0 100 3.9 90.9 2 3.2 0 100 0 0 0 32.5 67.5 0 0 0 100 0 3.8 0 94.9 1.2 100 0 0 0 100 0 0 0 36.3 8.8 29.5 25.5 11.1 11.1 11.1 66.7 19.8 10.5 69.6 0 24.8 12.7 46.6 15.9 12.5 36.7 49.2 1.5 31.8 12.6 39.7 16
LPM-style matrices look like this:
> letter-probability matrix V_STAT1_01: alength= 4 w= 21 nsites= 100.1 E= 0 0.080000 0.180000 0.419000 0.321000 0.232232 0.097097 0.372372 0.298298 0.138723 0.312375 0.197605 0.351297 0.175175 0.379379 0.381381 0.064064 0.595405 0.134865 0.150849 0.118881 0.311688 0.262737 0.093906 0.331668 0.000000 0.000000 0.000000 1.000000 0.000000 0.000000 0.000000 1.000000 0.039000 0.909000 0.020000 0.032000 0.000000 1.000000 0.000000 0.000000 0.000000 0.325000 0.675000 0.000000 0.000000 0.000000 1.000000 0.000000 0.038038 0.000000 0.949950 0.012012 1.000000 0.000000 0.000000 0.000000 1.000000 0.000000 0.000000 0.000000 0.362637 0.087912 0.294705 0.254745 0.111000 0.111000 0.111000 0.667000 0.198198 0.105105 0.696697 0.000000 0.248000 0.127000 0.466000 0.159000 0.125125 0.367367 0.492492 0.015015 0.317682 0.125874 0.396603 0.159840
References
-
Orenstein Y, and Shamir O.
PMID:24500199
A comparative analysis of transcription factor binding models learned from PBM, HT-SELEX and ChIP data
Nucleic Acid Research, 2014; 42(8):e63.
